

Búsqueda Simple
Búsqueda Completa
Taxonómica
Anatomía

+ Teoría
Patología
Aleatorio Principio Activo/ComercialFármacos o Drogas Más Visitadas
Levomepromazina

707943 visitas
Desvenlafaxina

274068 visitas
Clobenzorex
272559 visitas
Quetiapina

240712 visitas
Acepromazina

209102 visitas
Últimos comentarios


Sigue las noticias por


Total Fármacos Visitados
15.243.695
Desde Noviembre de 2008
CLASIFICACIÓN DROGAS


Nombre: Cocaína
Comercial: Coca, Perico, Manteca, Pasta, Merca, Sniff, Sniper
Foto:
Fórmula: 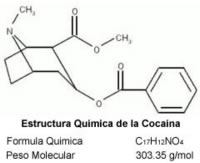
http://es.wikipedia.org/wiki/Erythroxylum_coca
Foto:
Categoría: Estimulante
Estado: Ilegal
Información: http://es.wikipedia.org/wiki/Cocaínahttp://www.drugabuse.gov/Infofacts/Cocaine-Sp.html
http://www.camporenacimiento.com/adiccion/cocaina.htm
Droga consultada 83389 veces
DROGAS DOPAMINÉRGICAS-NORADRENÉRGICAS
Feniletilaminas de síntesis
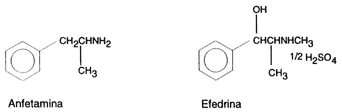
Drogas Anfetamínicas:
- Anfetamina y dextroanfetamina
- Metanfetamina (speed, ice, crystal, crystal meth)
- Efedrina (éxtasis verde, herbal ecstasy)
- Catinona y catina
- Metilfenidato y Pemolina (utilizados para el déficit de atención)
- Fenilpropanolamina (anorexígeno, descongestivo nasal)
- Anorexígenos
Las anfetaminas son un grupo de compuestos orgánicos del nitrógeno que pueden considerarse derivados del amoniaco. La amfetamina (C9H3CL), significa a (alfa), m(metil), f(fenil), et(il), amina
La precursora de la anfetamina es la efedrina y esta a su vez procede de la planta (Catha edulis) que ya había sido utilizada desde antiguo en el tratamiento del asma y a partir del descubrimiento de la efedrina se inció la aplicación terapéutica en determinadas enfermedades del sistema nervioso central.
Fuente:http://www.monografias.com/trabajos30/__/anfetaminas.shtml
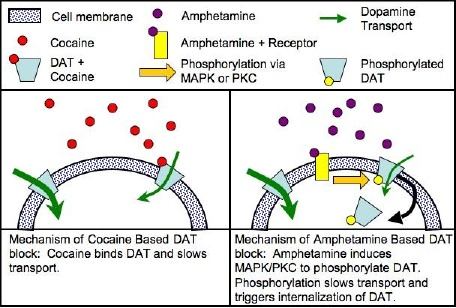
La cocaína se une directamente al transportador DAT1, inhibiendo la recaptación con más eficacia que las anfetaminas que fosforilan el DAT, causando la internalización del DAT (que la cocaína no lo hace) y sólo la inhibición de su recaptación como secundaria, es mucho menor el modo de acción que el de la cocaína, a partir de la conformación opuesto / orientación al DAT.
***
La metanfetamina está clasificada como un sicoestimulante al igual que otras drogas de abuso, como son la anfetamina y la cocaína. Sabemos que la estructura de la metanfetamina es similar a la anfetamina y el neurotransmisor dopamina pero que es muy diferente a la cocaína. Aunque estos estimulantes tienen efectos similares hacia el comportamiento y la fisiología, hay grandes diferencias en los mecanismos básicos de cómo trabajan al nivel celular del sistema nervioso. Sin embargo, la conclusión es que el resultado de la metanfetamina, tal como la cocaína, es la acumulación del neurotransmisor dopamina y esta concentración excesiva de la dopamina es la que aparentemente produce la estimulación y sensación de euforia que siente el usuario. En contraste a la cocaína, la cual se elimina rápidamente y es casi metabolizada por completo en el cuerpo, la metanfetamina tiene una duración de acción mucho más larga y un porcentaje mayor de la droga permanece sin cambiar en el cuerpo. El resultado es que la presencia de la metanfetamina en el cerebro dura más, lo cual finalmente conduce a la prolongación de los efectos estimulantes de la droga.
Núcleo Tropano

Coca y derivados: Cocaína
Su acción sobre la neurotransmisión dopaminérgica es el determinante primario de sus efectos reforzadores. Esta acción se produce sobre las neuronas dopaminérgicas procedentes del area tegmental ventral, que median las propiedades reforzadoras de las drogas de abuso. Se ha demostrado que la cocaína es una de las drogas con mayor capacidad de reforzamiento. Los primates y las ratas tienden a autoadministrarse la droga hasta quedar totalmente agotados. En humanos, se ha descrito que un adicto a la cocaína puede ilegar a inyectarse hasta veinticinco veces en un día. Los efectos secundarios que acompañan a su uso son psicosis, taquicardia, fallos cardiacos y el riesgo del SIDA, cuando se comparten las jeringuillas utilizadas para su inyección. La repetida administración de cocaína desarrolla tolerancia y sensibilización. La sensibilizacion puede jugar un importante papel en los ataques de pánico, en la paranoia y en la letalidad inducidos por la administración de cocaína. La sensibilización puede depender de un entorno determinado, como se observa en experimentos realizados con ratas, en las que el aumento de actividad motora debida a la droga es más bajo cuando se les cambia el lugar donde se realiza la inyección. El desarrollo de tolerancia pue de jugar un importante papel en el progresivo aumento de las dosis que necesitan los adictos.
***
La farmacodinamia de la cocaína involucran las complejas relaciones de los neurotransmisores (inhibiendo la captación de monoaminas en ratas con una relación cercana a: serotonina:dopamina = 2:3 serotonina:noradrenalina = 2:5 [54]) El efecto más estudiado de la cocaína en el centro sistema nervioso es el bloqueo de la proteína transportadora de dopamina. el transmisor dopamina liberado durante la señalización neuronal es normalmente reciclado a través del transportador, es decir, el transportador se une el transmisor y lo bombea hacia fuera de la hendidura sináptica de nuevo en la neurona presináptica, en el que se recoge en las vesículas de almacenamiento. La cocaína se une fuertemente al transportador de dopamina formando un complejo que bloquea la función de transportista. El transportador de la dopamina no puede seguir desempeñando su función de la recaptación de la dopamina y por lo tanto se acumula en la hendidura sináptica. Esto resulta en un efecto postsináptico aumentado y una prolongación de la señalización dopaminérgica en los receptores de dopamina en la neurona receptora.
La exposición prolongada a la cocaína, como ocurre con el uso habitual, conduce a la desregulación de la homeostasis de la normalidad (es decir, sin la cocaína) de la señalización dopaminérgica a través de la baja regulación de los receptores de la dopamina y la transducción de la señal. La disminución de la señalización dopaminérgica después del uso crónico de cocaína puede contribuir a trastornos del estado de ánimo depresivo y sensibilizar a este circuito de recompensa del cerebro importante para los efectos reforzantes de la cocaína (por ejemplo, mejorar la señalización dopaminérgica sólo cuando la cocaína es auto-administrada). Este efecto contribuye a la naturaleza intratable de la adicción y la recaída.
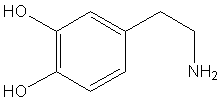
Estructura química de la dopamina (C6H3(OH)2-CH2-CH2-NH2)
Junto con los opiáceos y el alcohol, la cocaína es la droga de abuso de la que más datos moleculares disponemos. Tras su inyección aguda parece inhibir la recaptación de catecolaminas y de serotonina. Su unión al trans-portador de dopamina produce un cambio conformacional en éste, que disminuye su afinidad por dopamina, con lo que disminuye su recaptación.
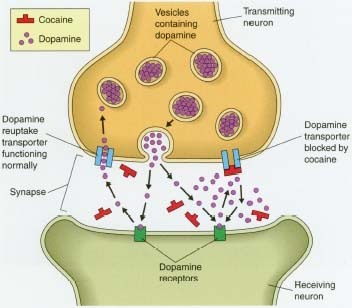
El aumento de dopamina en el espacio intersináptico produce diversos efectos como pueden ser la inhibición de la síntesis de dopamina o la inhibición de la liberación de neurotransmisores por parte de las células postsipnápticas en caudado, núcleo accumbens y corteza prefrontal medial. La inhibición de la recaptación de serotonina en el rafe dorsal y de norepinefrina en el locus cerúleo pueden ser los responsables de la inhibición de la actividad neuronal que se produce en dichas áreas. El efecto de la cocaína sobre el transportador de norepinefrina puede contribuir a algunas de SUS acciones nocivas sobre el sistema cardiovascular, que incluso pueden llegar a ser letales.
El bloqueo del receptor de serotonina puede tener un efecto sobre las células dopaminérgicas del area tegmental ventral, dado que este area recibe neuronas serotoninérgicas procedentes de los núcleos de Rafe. El que estas acciones de la cocaína se realicen sobre varios transportadores de monoaminas puede explicar por qué los bloqueantes específicos del transporte de dopamina son incapaces de mimetizar todos los efectos producidos por esta droga.
Fuentes:
- http://en.wikipedia.org/wiki/Cocaine
- http://www.biografica.info/redei/neurobiologia-de-la-drogradiccion
- http://www.analisisdedrogas.es/cocaina_13.html
- http://en.wikipedia.org/wiki/Substituted_amphetamine
Mecanismo de acción del Transportador de recaptación de la dopamina DAT1
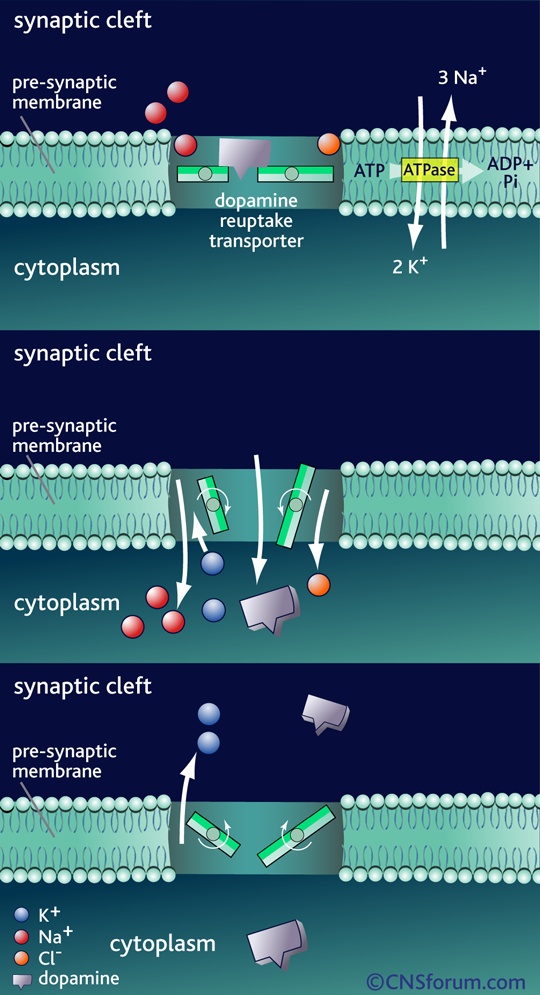
La acción de la dopamina en la sinapsis se termina con su recaptación por la membrana presináptica. Este es un proceso dependiente de energía. Sodio/potasio ATPasa usa la energía de hidrólisis del ATP para crear un gradiente de concentración de iones a través de la membrana presináptica que impulsa la apertura de los transportistas y el co-transporte de iones sodio y cloruro junto con la dopamina a la hendidura sináptica. Los iones de potasio son neesarios para que el transportista pueda volver a la posición externa. Con la liberación de los iones de potasio en la hendidura sináptica se equilibra el gradiente de iones a través de la membrana presináptica. El transportador de la recaptación de dopamina está entonces disponible para unirse a otra molécula de la dopamina para la re-absorción.
Los transportadores de membrana de dopamina y serotonina tienen mucho en común, constando en ambos casos de 12 dominios transmembrana que transportan uno o dos iones de sodio y un ión de cloruro con cada molécula de neurotransmisor. Se encuentran en la terminal presináptica, donde recaptan los neurotransmisores mediante un sistema de alta afinidad dependiente de sodio y cloruro, impulsado por una bomba sodio-potasio ATPasa.
El transportador de membrana de dopamina DAT1 (del inglés, Dopamine Transporter 1) es el punto de acción de las sustancias estimulantes de abuso, como la cocaína y las anfetaminas.
Fuentes:
- http://en.wikipedia.org/wiki/Dopamine_transporter
- http://www.cnsforum.com/imagebank/item/rcpt_sys_DA_reup/default.aspx
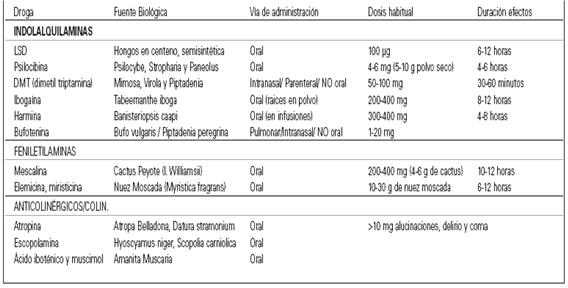
DROGAS SEROTONINÉRGICAS
ALUCINÓGENAS INDOLES
Alcaloides de anillo indólico
Indolaminas
Drogas Alucinógenas Indoles:
La estructura química de gran parte de sustancias alucinógenas lleva un núcleo que es un anillo indol. Se trata de una sustancia heterocíclica formada por la unión del benceno y del pirrol. Es, junto con el escatol (beta-metilindol), uno de los productos de la putrefacción de las proteínas y está presente en el excremento del animal. Su nombre de indol se debe a que se encuentra en el colorante índigo. Debido a los dos átomos de N que además llevan en la cadena lateral, la estructura química de estos alucinógenos indólicos corresponde a la fundamental de la triptamina.
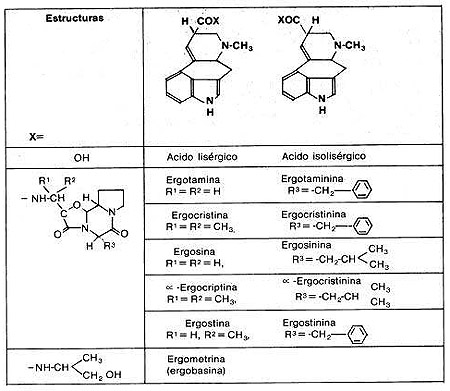
Ergotaminoides: LSD, isoergida
Triptaminas:DMT, psilocibina, psilocina, bufotenina
Carbolinas: harmina, harmalina (Son inhibidores de la monoaminoxidasa (IMAOs) reversibles no son alucinógenos, que permiten que se active oralmente la DMT), ibogaína
Por ejemplo, el LSD inhibe la liberación de serotonina al unirse a los receptores presinápticos de este receptor. La consiguiente inhibición de la actividad se rotoninérgica produce la desinhibición de otras rutas bajo su control. El que la serotonina esté implicada en el control de las sensaciones, el sueño, la atención y el estado de ánimo, podría explicar la acción del LSD y los otros alucinógenos sobre estas críticas funciones cerebrales. También se ha descrito que el LSD y las anfetaminas metiladas actúan sobre las neuronas dopaminérgicas. Desde un punto de vista químico todas las drogas psicodélicas más importantes guardan un estrecho parecido con los neurotransmisores serotonina, norepinefrina o dopamina. Dos de los anillos del LSD son idénticos al anillo de indol de la serotonina, y la cadena lateral de esta última es idéntica a otra parte del LSD. La estructura de la mescalina se parece más a la de norepinefrina o a la de dopamina. Psilocina, psilocibina y dimetilitriptamina son muy parecidas a la serotonina.
Fuentes: http://www.biografica.info/redei/neurobiologia-de-la-drogradiccion
LSD afecta a un gran número de los receptores acoplados a proteínas G, incluyendo todos los subtipos de receptores de dopamina, y todos los subtipos de receptores adrenérgicos, así como muchos otros. El LSD se une a la mayoría de los subtipos del receptor de serotonina, excepto para el 5-HT3 y 5-HT 4. Sin embargo, la mayoría de estos receptores se ven afectados en dosis muy bajas en afinidad, es lo suficientemente activa con una concentración cerebral de aproximadamente 10 a 20 nm. [82] En los seres humanos, las dosis de LSD recreativas pueden afectar a: 5-HT 1A, 5-HT 2A, 5-HT 2C, 5-HT 5A y otros 6 receptores 5-HT. [1] [83] El subtipo 5B receptores de 5-HT, que no están presentes en los seres humanos, también tienen una alta afinidad por el LSD. [84] Los efectos psicodélicos del LSD se atribuyen a sus fuertes efectos de agonista parcial del 5-HT 2A receptores específicos como los agonistas 5-HT 2A son psicodélicos y en gran medida los antagonistas del 5-HT 2A bloquean la actividad psicodélica del LSD. [82] Exactamente cómo estos efectos de la droga se producen se desconoce, pero se cree que actúa aumentando la liberación de glutamato en la corteza cerebral y por lo tanto la excitación en esta área, específicamente en las capas IV y V. [85] LSD, al igual que muchos otras drogas, se ha demostrado que activan las vías DARPP-32-relacionadas. [86]
Ver afinidad del LSD a los receptores según constante de inhibición (Ki) expresada en nanomoles.
LSD y alucinógenos
http://www.adicciones.es/files/(10)%20sole.pdf
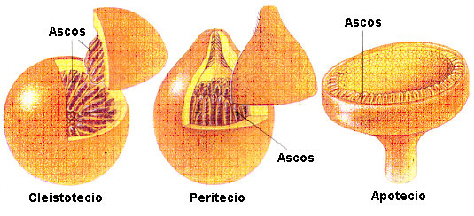
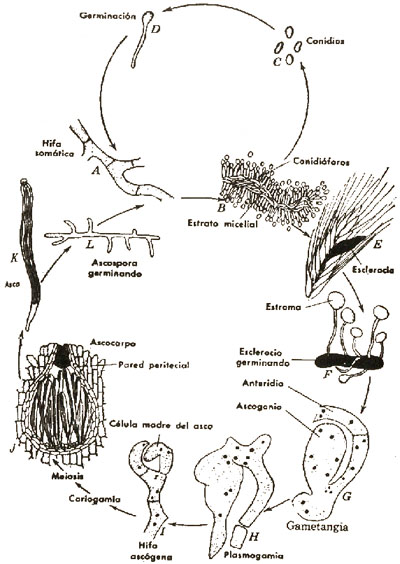
 Cornezuelo de Centeno
Cornezuelo de Centeno

ENTACTÓGENAS
Feniletilaminas de síntesis
Análogos a las anfetaminas
Anillo bencénico
Drogas Entactógenas:
Se ha sugerido que la MDMA y las sustancias que poseen efectos psico-farmacológicos semejantes a los de esta droga, son miembros de una misma familia farmacológica, denominados entactógenos. Se pueden definir los entactógenos como sustancias que al tener efectos empáticos, facilitando las relaciones interpersonales, son capaces de facilitar el acceso al interior de la conciencia del individuo, para un mejor control de los conflictos emocionales, pareciendo especialmente útil esta propiedad en terapias psicoanalíticas. Los fármacos entactógenos son distintos a los denominados estimulantes centrales (prototipo anfetamina) o a los alucinógenos (prototipo la anfetamina metoxilada DOM). Esta diferencia ha sido puesta de manifiesto en estudios de radioelectroencefalografía, de relación estructura-actividad y por diferencias bioquímicas.
Anfetaminas entactógenas (derivados metilenodioxianfetaminas)
- 3,4-metilenodioxianfetamina (MDA, "pildora del amor") Metabolito del MDMA, más alucinógena que entactógena
- 3,4-metilelenodioximetanfetamina (MDMA, "éxtasis", "Adán", XTC) Más entactógena
- 3,4-metilenodioxietilanfetamina (MDEA o MDE, "Eva") Menos potente, menos depleción de Serotonina
- N-metil-1-(3,4-metilenodioxifenil)-2 butamina (MBDB). Análogo al MDMA
Al igual que con otras anfetaminas, la MDMA produce la liberación de estos neurotransmisores de sus sitios de almacenamiento dentro de las neuronas, lo que resulta en una mayor actividad neurotransmisora. En comparación con el poderoso estimulante metanfetamina, la MDMA produce una mayor liberación de serotonina y una menor de dopamina. La serotonina es un neurotransmisor que juega un papel importante en la regulación del estado de ánimo, sueño, dolor, emociones, apetito y otros comportamientos. La liberación excesiva de serotonina causada por la MDMA probablemente produce los efectos de elevación en el estado de ánimo que sienten los usuarios de esta droga. Sin embargo, al liberar cantidades grandes de serotonina, la MDMA disminuye significativamente las cantidades de este importante neurotransmisor en el cerebro, contribuyendo así a los efectos negativos posteriores en el comportamiento que los usuarios frecuentemente experimentan por varios días después de haber tomado MDMA.
***
Vamos a describir el del MDMA, dado que disponemos de muchos datos sobre cómo altera la funcionalidad cerebral. Actúa como agonista indirecto de serotonina, facilitando su liberación e inhibiendo su recaptación, lo que aumenta la presencia del neurotransmisor en dicho espacio intersináptico. Modula la acción de la serotonina liberada al unirse a los receptores serotoninérgicos 5HT2, que parecen ser de gran importancia para la actuación de las drogas alucinógenas clásicas como el LSD. Su unión a receptores adrenérgicos a2, puede estar relacionada con algunos de los eiectos cardiovasculares atribuidos a la droga.
El MDMA inhibe la liberación de dopamina en las neuronas dopaminérgicas de la vía nigroestriatal, de una forma indirecta a través de la liberación local de serotonina. Este efecto podría estar relacionado con los efectos estimulantes asociados al MDMA.
Aunque no está claro si el MDMA es adictivo, la deplección de los almacenes de serotonina puede conducir a depresión y anhedonia, y estos sentimientos pueden aumentar su consumo. No se conocen las consecuencias a largo plazo de su uso, pero existen diversos estudios con animales que podrían relacionarse con las alteraciones comportamentales observadas en sus consumidores. Se ha visto una degeneración de algunas neuronas serotoninergicas en roedores y en primates no humanos después de su administración prolongada. El mecanismo propuesto para explicar cómo puede el MDMA alterar las terminales serotoninérgicas podría ser similar al que ha sido postulado para la destrucción de las terminales dopaminérgicas en la enfermedad de Parkinson y que en la forma postulada por M. Rattray aparece recogida en la figura 6.
El MDMA se metaboliza a una estructura quinoide (el aducto con glutacion de dihidroximetanfetamina) que puede entrar en las células nerviosas. Diversos procesos redox en los que participa este metabolito pueden producir radicales libres de oxígeno, que inactivarían la triptóíano hidroxilasa y causarían daños a los componentes proteicos y lipídicos de la terminal presináptica.
La unión del MDMA a los receptores serotoninérgicos 5HT2 podría incrementar la concentración de calcio citoplasmático, que entre otras acciones podría incluir proteolisis y estrés osmótico, que también contribuirían a la degeneración celular. Esta pérdida neuronal junto con la disminución de los niveles de serotonina pueden ser responsables de la aparición de enfermedades neurosiquiátricas a largo plazo, un ejemplo de las cuales podrían ser los casos recientemente descritos entre sus consumidores de psicosis paranoide crónica.
Fuente:
- http://www.biografica.info/redei/neurobiologia-de-la-drogradiccion-
- http://www.analisisdedrogas.es/mdma_21.html
Con propiedades mixtas estimulantes y alucinógenas de las anfetaminas y la mescalina
Anfetaminas alucinógenas (derivados metoxianfetaminas):
- 4-bromo-2,5-dimetoxianfetamina ((DOB) 100 veces más potente que las anfetaminas
- 4-metil-2,5-dimetoxianfetamina (DOM, serenity-tranquility-peace o STP) Mayores propiedades Alucinógenas
- 2,4,5-trimetoxianfetamina (TMA-2) Análogo a la Mescalina
- Parametoxianfetamina (PMA) Análogo al LSD
Alucinógenos clásicos
DROGAS ANTICOLINÉRGICAS
Alcaloides tropanos
Alucinógenos clásicos:
AGENTES CON EFECTOS ANTICOLINÉRGICOS
En este grupo se encuentran algunas de las plantas con mayor tradición e historia escrita, célebres por su papel en la leyenda y la literatura de varias culturas europeas, hindúes y americanas. Pertenecen a la misma familia que la papa, las Solanáceas. Tres de ellas se han empleado básicamente en Europa e India: la Atropa belladona, la Mandragora officinarum y el Hyoscyamus niger (beleño). La cuarta, de la especie Datura, se distribuye mundialmente (nuestro famoso toloache).
La belladona es célebre por las historias de envenenamientos, brujería y ceremonias secretas asociadas a su uso. En Las mil y una noches se describe una muerte con veneno (probablemente atropina); hay historias medievales que sugieren su consumo en misas negras, incluyendo las narraciones de brujas que vuelan en escobas; la mandrágora se describe en la Biblia y el beleño es mencionado por Plinio, en el año 60 d. C. Diferentes especies de Datura también se han conocido desde hace siglos: entre los chinos se le ha asociado a Buda; entre los antiguos griegos se le utilizaba en ceremonias adivinatorias y quizá ritos dionisiacos; entre los hindúes se usaba en ritos ligados a Shiva; y en Mesoamérica, Francisco Hernández, protomédico real de Felipe II, la describe en su famosa Farmacopea (¿y quién no ha escuchado alguna historia de hechicería realizada mediante la administración de toloache?)
Dada la amplia distribución central y periférica de este receptor (véase el capítulo V), los efectos de estos alcaloides ocurren a muy diversos niveles: sequedad de la boca, taquicardia, aumento de la temperatura corporal, disminución del peristaltismo gastrointestinal (p. ejem., constipación), dilatación pupilar, confusión mental, obnubilación de la consciencia, pérdida de la memoria reciente, y somnolencia, delirio y coma a dosis elevadas. A diferencia de otros alucinógenos, los anticolinérgicos no incrementan la percepción sensorial.
Fuente: http://omega.ilce.edu.mx:3000/__//html/sec_29.html
DROGAS DISOCIATIVAS
(Anestésicas)
GLUTAMATÉRGICAS
Arilciclohexilaminas
Análogos a la fenciclidina
Drogas Disociativas Anti-NMDA:
Psicosis inducida por PCP, de glutamato a GABA
El mecanismo a través del cual dosis bajas de antagonistas de receptores NMDA llevan a un aumento en la liberación de glutamato en la corteza cerebral, permanecía en el misterio hasta que un reciente estudio mostrara que la administración sistémica y no local de PCP en la corteza prefrontal induce hiperactividad eléctrica prefrontal en animales de laboratorio, sugiriendo así que acciones de esta droga en regiones ajenas a la corteza prefrontal son necesarias para la inducción de estados hiperglutamatérgicos prefrontales. Buscando establecer la región del cerebro involucrada en este fenómeno, los investigadores inyectaron localmente PCP en el hipocampo ventral, una región que envía masivas proyecciones glutamatérgicas a la corteza prefrontal. Así, pudieron demostrar que la hipofunción de receptores NMDA expresados por neuronas localizadas en esta región del cerebro conducía a una actividad eléctrica prefrontal aumentada (61). Con respecto a la identidad de estas neuronas, estudios previos habían mostrado que un sub-grupo de interneuronas GABAérgicas del hipocampo son particularmente sensibles al antagonismo NMDA(62). La causa de esta hipersensibilidad se debería a que estas interneuronas expresan el sub-tipo NR2D del receptor NMDA, el cual, difiriendo de aquellos expresados por neuronas glutamatérgicas, es insensible al bloqueo por Mg++. Dado que PCP y otros antagonistas NMDA de su tipo, sólo bloquean aquellos receptores NMDA que se encuentran abiertos, es decir aquellos que han sido liberados del bloqueo de Mg++ por actividad eléctrica concomitante, o que por razones estructurales son insensibles a ese bloqueo, las interneuronas que expresan NR2D resultan 10 veces más sensibles que las neuronas glutamatérgicas al bloqueo de sus receptores NMDA(63). Así, la hipoactividad de estas interneuronas llevaría a su vez a la desinhibición de las neuronas glutamatérgicas que proyectan a la corteza prefrontal. De esta manera, la inducción de síntomas psicóticos, así como déficit cognitivos y emocionales en sujetos agudamente tratados con antagonistas de receptores NMDA sería mediada por inactivación de interneuronas GABA. Confirmando la validez de este modelo para la comprensión de la esquizofrenia, el hallazgo mejor replicado en la neuropatología de esta enfermedad es la presencia de marcadores moleculares sugiriendo déficit en la actividad de interneuronas GABAérgicas en diversas regiones del cerebro, tales como corteza prefrontal, corteza temporal, hipocampo y cerebelo(64). Más aún, un estudio reciente ha reportado que poliformismos en la región 5' del gen que codifica para GAD- 67, la enzima más importante en la síntesis de GABA en el cerebro, predicen la magnitud de la disminución progresiva en sustancia gris observada en pacientes con formas de psicosis esquizofrénica de inicio tanto en la infancia como en la vida adulta(65). Surge entonces la pregunta sobre el mecanismo a través del cual la hiperactividad glutamatérgica inducida por déficit en la actividad de interneuronas GABAérgicas podría llevar a disminución en la sustancia gris de pacientes esquizofrénicos en las fases incipientes de esta enfermedad.
Fenciclidina:
Farmacodinamia
El alcance de la farmacología de este compuesto en el sistema nervioso central humano no está completamente entendido, sino que se une a muchos receptores diferentes. Las interacciones principales son como un antagonista no competitivo de la subunidad-3A [subunidad épsilon] del receptor de NMDA en el Homo sapiens. La fenciclidina se sabe que se une con alta afinidad, a la subunidad D1 de la persona humana DAT (transportadores de dopamina), además de mostrar un efecto positivo antagónico en el ?7 subunidad del receptor nicotínico de acetilcolina (nAChR). También se une al receptor mu-opioide, lo que parece ser una parte central del mecanismo de acción de los fármacos en esta clase.
http://en.wikipedia.org/wiki/Phencyclidine
Ketamina:
Farmacodinamia:
La ketamina mucho tiempo se pensó que actúa principalmente mediante la inhibición de los receptores NMDA. [53] [54] Sin embargo, otro antagonista del receptor NMDA, MK-801, no ejerce efectos hipnóticos de la misma. [55] Parece más probable que los efectos hipnóticos de la ketamina son producidos por la inhibición de canales activados por hiperpolarización modulados por nucleótidos cíclicos de cationes (HCN1) que median el "hundimiento" de la corriente activada por hiperpolarización (Ih) en las neuronas.[56] La ??inhibición de la Ih por ketamina en cultivos de neuronas provoca un cambio hiperpolarizante en reposo y aumenta el potencial de membrana sumada a las corrientes de excitación. [57] Estos efectos, si son inducidos en vivo, probablemente inducirían oscilaciones corticales que recuerdan a dormir. [58] lo que es más importante, sin los HCN1 en ratones se elimina las acciones hipnóticas de la ketamina.
La ketamina es un antagonista del receptor NMDA no competitivo. [59] Este receptor se abre en respuesta a la unión del neurotransmisor glutamato, y el bloqueo de estos receptores se cree que median la analgesia (reducción del dolor) los efectos de la ketamina en dosis bajas. [54] Evidencia para esto se refuerza por el hecho de que la naloxona, un antagonista de los opiáceos, no revierte la analgesia. Los estudios también parecen indicar que la ketamina es de "uso dependiente" lo que significa que sólo inicia su acción de bloqueo una vez que el glutamato se une al receptor NMDA. En dosis altas, el nivel de anestesia completa, la ketamina también se ha encontrado para unirse a los receptores opioides mu y receptores sigma. Por lo tanto, la pérdida de la conciencia que se produce en altas dosis puede ser parcialmente debido a la unión a receptores de opioide mu y sigma . También se ha demostrado que actúa como un potente agonista de los receptores D2 parcial, [60], así como un inhibidor de la recaptación de la dopamina. (RAC), la ketamina es un inhibidor no competitivo de la nAChR ?7 a concentraciones clínicamente relevantes.
- http://en.wikipedia.org/wiki/Ketamine
- http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2744993/?tool=pmcentrez
- http://www.revespcardiol.org/es/revistas/revista-espa%C3%B1ola-cardiologia (importante)
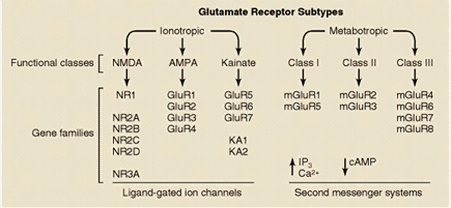
A diferencia de los otros dos tipos de receptores ionotrópicos, AMPA y kainato, el receptor NMDA posee una serie de características distintivas que lo hacen único entre todos los receptores ionotrópicos. Uno de esos aspectos, quizá el más significativo, es que el canal formado por el receptor permite el paso de los iones Ca2+, además del Na+ y K+, lo que implica un incremento de la concentración de Ca2+ intracelular en la neurona postsináptica cada vez que el receptor se activa.
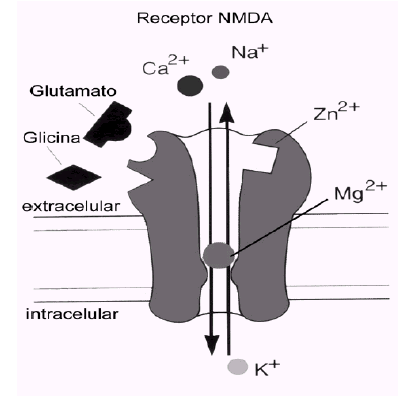
El receptor NMDA es una proteína muy compleja y tremendamente regulada (ver figura). Su conductancia al Ca2+ es notablemente alta y es ésta quizá su característica más destacable y la responsable de muchas de sus funciones. Otra característica especial del receptor NMDA es que para que el canal se abra se necesita, además del glutamato, la presencia de un co-agonista (el aminoácido glicina). Ciertas poliaminas, al igual que la glicina, modulan positivamente el canal, mientras que el cinc y un exceso de protones lo modulan negativamente. Sin embargo, lo más llamativo de este receptor es que comparte características funcionales de canales regulados por ligando y de canales sensibles al voltaje y dependientes de uso. Esta propiedad está relacionada con el bloqueo efectivo del canal del receptor NMDA por el ion Mg2+, cuando el potencial de membrana está próximo al valor de reposo. Este bloqueo es eliminado transitoriamente cuando la membrana se despolariza, por estimulación repetitiva previa, por ejemplo.
Los receptores NMDA son complejos proteicos formados por diferentes combinaciones de varias subunidades (denominadas NMDAR1 y NMDAR2A-2D). La subunidad NMDAR1 posee todas las propiedades fundamentales necesarias para constituir un canal funcional y puede estar presente en ocho isoformas diferentes. La otra familia de proteínas que contribuye a la formación de receptores NMDA funcionales está constituida por cuatro variantes de la subunidad NMDAR2 (NMDAR2A-2D), codificadas por cuatro genes separados. Distintas combinaciones de la subunidad fundamental NMDAR1 con las otras subunidades dan lugar a receptores NMDA con propiedades funcionales diferentes, que pueden estar distribuidas en áreas encefálicas específicas y/o que pueden definir respuestas fisiológicas o patológicas distintas en respuesta al glutamato.
Fuentes:
- http://www.encuentros.uma.es/encuentros83/nmda.html
- http://www.monografias.com/trabajos30/receptor-nmda/receptor-nmda.shtml
- http://www.scielo.org.ve/scielo.php?pid=S0798-02642004000200002&script=sci_arttext
- http://www.javeriana.edu.co/Facultades/Ciencias/neurobioquimica/libros/neurobioquimica/glutamaspatato.htm
- http://www.ciencia.cl/CienciaAlDia/volumen5/numero2/articulos/articulo5.html
Una gran parte de las acciones mediadas por los receptores NMDA se basa en la regulación del flujo de Ca2+ hacia el interior de la célula. La activación de los receptores NMDA permitiría un rápido influjo de Ca2+, con la consiguiente elevación intracelular de Ca2+, lo cual dispararía una cascada de sistemas de segundos mensajeros que podría producir acciones muy diversas.
Glutamato y receptores NMDA están involucrados en numerosas funciones dentro del sistema nervioso. Uno de los procesos más estudiados en el que los receptores NMDA parecen juegar un papel clave es la plasticidad sináptica. La maduración de los circuitos nerviosos (establecimiento de conexiones funcionales) durante el desarrollo, y también en el adulto, depende de la activación y consolidación de ciertas sinapsis, mediante mecanismos de plasticidad en el que están involucrados los receptores NMDA. La potenciación a largo plazo (LTP), una forma de plasticidad sináptica que está en la base de los procesos de aprendizaje y memoria, implica la activación de los receptores NMDA. También ha sido demostrado recientemente un papel crucial de los receptores NMDA en los procesos de formación de las memorias, incluida la denominada memoria episódica, un tipo de memoria que nos permite recordar las experiencias vividas, aunque los acontecimientos solamente ocurran una vez. Otros estudios han demostrado un papel del glutamato a través de su unión con receptores NMDA en los procesos de emigración celular.
DROGAS COLINÉRGICAS
Alcaloides de núcleo pirídico
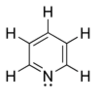

La acetilcolina (AC) fue el primer neurotransmisor caracterizado tanto en el sistema nervioso periférico (SNP) como en el sistema nervioso central (SNC) de los mamíferos, el cual participa en la regulación de diversas funciones como fenómenos de activación cortical, el paso de sueño a vigilia y procesos de memoria y asociación. La AC se sintetiza a partir de la colina y del acetil CoA, en una reacción catalizada por la colina acetiltranferasa (CAT) y existen mecanismos que regulan de manera precisa su síntesis y liberación. Las técnicas de clonación molecular han permitido la identificación de dos tipos de receptores: ionotrópicos (nicotínicos) y metabotrópicos (muscarínicos)estos acoplados a proteínas G. Los receptores M1, M3 y M5 están acoplados a la activación de proteínas Gq, con la consecuente activación de la fosfolipasa C. Los receptores M2 y M4 inhiben la formación de AMPc, activan canales de K+ y reducen la entrada de iones de Ca++ a través de canales dependientes del voltaje, efectos mediados por proteínas G (Gai y Gao). Los receptores de acetilcolina se encuentran ampliamente distribuidos en diversas áreas del SNC y en el SNP, en donde cada uno de ellos presenta un patrón de expresión temporal y espacial particular, los cuales pueden sobreponerse durante el desarrollo y son responsables de las diversas acciones fisiológicas de la acetilcolina. El estudio de los sistemas y receptores colinérgicos del SNC ha generado gran interés, debido a que diversas alteraciones en la transmisión colinérgica han sido relacionadas, directa o indirectamente, con trastornos severos como la enfermedad de Alzheimer y la de Parkinson.
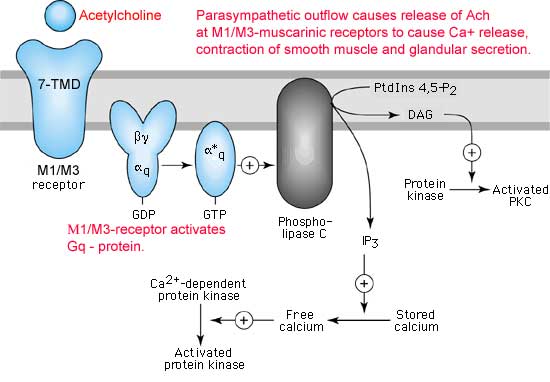
Los receptores nicotínicos para la acetilcolina han sido aislados, purificados, clonados y secuenciados. Están formados por cuatro subunidades (llamadas a, b, g y d) cuyas secuencias son conocidas y muestran un 35-40% de homología. Cada una de ellas, atraviesa la membrana cinco veces a través de a-hélices M1 a M5 (*). El receptor completo contiene dos subunidades a (donde se fija la acetilcolina) y una subunidad de cada uno de los tres subtipos restantes (*). Al fijarse la acetilcolina al receptor, se abre un canal central por donde puede entrar sodio en la célula.
La estructura y función de los receptores muscarínicos colinérgicos son muy distintas de las de los nicotínicos. Se han identificado al menos 5 subtipos de receptores muscarínicos. Los M1 y M2 están formados por 7 segmentos transmembranarios y ejercen sus acciones a través de proteínas G. La activación de M1 ocasiona una disminución de la conductancia para el K+ por medio de la activación de una fosfolipasas C, mientras que la activación de los receptores M2 produce un aumento de la conductancia del K+ por medio de la inhibición de la adenilciclasa. La unión de la acetilcolina a los M1 produce pués una depolarización de la membrana postsináptica mientras que la unión de la acetilcolina a los M2 produce una hiperpolarización
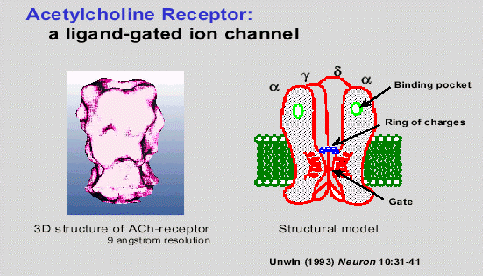
Esquema cíclico de funcionamiento del receptor nicotínico de la acetilcolina
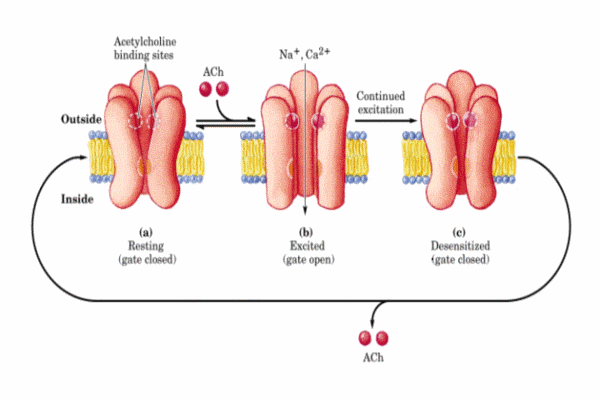
Se conocen diversas sustancias que competen con la acetilcolina en estos receptores. La a-bungarotoxina (una toxina del veneno de una serpiente del sudeste asiático) se fija específicamente e irreversiblemente al receptor bloqueando su acción. Lo mismo ocurre con la toxina botulínica. Otros fármacos antagonistas de la acetilcolina son la nicotina, la tubucurarina, etc. Todos ellos, inducen una serie de síntomas clínicos (relajación muscular que puede llegar a un bloqueo completo, hipotensión, bradicardia, etc). Para restaurar una membrana depolarizada a su estado excitable, el necesario eliminar o destruir la señal depolarizante. Existen tres formas de terminar la señal: * el transmisor difunde fuera del hendidura sináptica * el transmisor es capturado por la neurona presináptica (proceso denominado recaptación) * el transmisor en degragado enzimáticamente En el caso de la acetilcolina, la enzima acetilcolinesterasa presente en la hendidura sináptica y en la membrana de la célula degrada la acetilcolina a colina y acetato.
Algunas neurotoxinas y gases nerviosos son inhibidores de la colinesterasa. Al prolongar los efectos de la acetilcolina, incrementa el tiempo durante el cual la membrana se encuentra despolarizada impidiendo la relajación de los músculos, en particular de los músculos respiratorios, ocasionando la muerte.
La miastenia grave es un desorden autoinmune caracterizado por debilidad muscular debido a un déficit de la transmisión neuromuscular, en la que está implicada la acetilcolina. Los pacientes con miastenia grave muestran anticuerpos a los receptores colinérgicos nicotínicos. Se cree que estos anticuerpos reaccionan con el receptor inhibiendo su función, ya sea su capacidad para captar la acetilcolina, ya sea no experimentando los cambios de conformación que permiten la entrada del sodio.
Fuentes:
- http://www.iqb.es/cbasicas/bioquim/cap9/c9s01_31.htm
- http://es.wikipedia.org/wiki/Receptor_de_acetilcolina
Drogas Colinérgicas:
Receptor Ionotrópico Nicotínico (nAChR):
Receptor Metabotrópico Muscarínico (mAChR):
La nicotina se absorbe por la piel y por la mucosa de la boca y la nariz o se inhala a través de los pulmones. La molécula alcanza pronto el cerebro del fumador. Al inhalar, el humo hace llegar la nicotina a los pulmones, con las partículas de alquitrán asociadas; de ahí, pasa a la sangre. De entre diez a sesenta segundos después, la nicotina atraviesa la barrera hematoencefálica y penetra en el cerebro.
Cuando no se inhala el humo, la nicotina se absorbe más lentamente a través de las membranas mucosas de la boca.
De los aproximadamente 3000 productos que contiene el cigarrillo, solo la nicotina crea dependencia. Su efecto es funesto en el segmento ventral del mesencéfalo y en el nucleus accumbens del prosencéfalo, en las áreas que forman parte del sistema de recompensa. La nicotina se vincula aquí a los receptores nicotínicos de la acetilcolina (nAChR) de las neuronas. Imita al neurotransmisor acetilcolina, que suele acoplarse a esas proteínas canaliculares y, de ese modo, cuida de que las neuronas liberen abundante dopamina.[1]
El influjo de la nicotina en las sinapsis dura varios minutos promoviendo una excitación persistente de las neuronas involucradas, efecto que se debilitará cuando la sensibilidad por exceso haga acto de presencia.
La causa de que la nicotina cree adicción está en que, aunque inicialmente las neuronas gabaérgicas a las que se acopla liberan el neurotransmisor ácido gammaaminobutírico que controla la liberación de dopamina en las neuronas vecinas, si aquellas son sobre-nicotinizadas, entonces la secreción del neurotransmisor se limita, con lo que la excitación dopamínica de estas otras neuronas aumenta.
La consecuencia de lo anterior a largo plazo es que las células adaptan su bioquímica, y esto ocurre en dos fases: primero crecen los receptores de nicotina, por lo que aumenta la secreción de dopamina; sin embargo, con el tiempo las neuronas reaccionan de forma menos inmediata a la nicotina, por lo que las necesidades de ingerir más cantidad aumentan.
***
El mecanismo de acción de la nicotina a nivel molecular está claramente definido. Es un agonista del receptor nicotínico de acetilcolina en el sistema nervioso periférico y en el central.
La acción estimuladora de la nicotina sobre el cerebro parece ejercerse sobre las neuronas noradrenérgicas procedentes del locus cerúleo y sobre las dopaminérgicas del area tegmental ventral. El locus cerúleo parece jugar un papel crucial en la vigilancia y en el despertar, en las reacciones relacionadas con el estrés y en la regulación de la actividad psicosomática. La administración tanto aguda como crónica de nicotina induce la actuación de esta región cerebral. Esto explica la activación de las reacciones de atención asociadas a su ingesta. Esta acción sobre el núcleo cerúleo podría explicar los efectos favorables de la clonidina en la abstinencia al tabaco, dado que, como ya se indicó en el capítulo tercero, este fármaco es un agonista 2-adrenérgico.
La actuación de la nicotina sobre el área tegmental ventral produce una estimulación motora y da lugar a un aumento de la liberación de dopamina en el núcleo accumbens. Esta capacidad de incrementar los niveles extracelulares de dopamina es una característica que la nicotina comparte con otras drogas de abuso como la cocaína, los opiáceos y el alcohol, y está relacionada con los sistemas de reforzamiento. Esto justificaría la conducta de búsqueda de droga asociada a los consumidores de tabaco, pese al conocimiento del peligro asociado a su uso.
Fuentes:
- http://www.biografica.info/redei/neurobiologia-de-la-drogradiccion
- http://www.genomasur.com/lecturas/Guia07.htm>
- http://www.jpp.krakow.pl/journal/archive/03_09/articles/01_article.html
- http://es.wikipedia.org/wiki/Receptor_de_acetilcolina
DROGAS GABAÉRGICAS
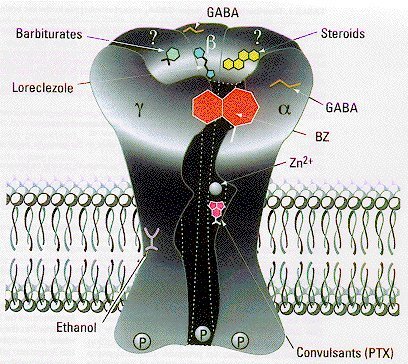
Drogas Gabaérgicas:
Receptor GABA-A:
Acción sobre el glutamato-nmda
Diversos estudios farmacológicos han demostrado que el alcohol a corto plazo aumenta las acciones GABA (neurotransmisor inhibitorio) sobre sus receptores y disminuye las de los aminoácidos excitatorios, tales como el glutamato, sobre los receptores NMDA.
Simplificando, podemos decir que el alcohol reduce la frecuencia de los impulsos eléctricos y deprime la actividad del SNC.
Cuando la exposición al alcohol es crónica, el organismo se adapta disminuyendo la actividad del sistema Gabaérgico inhibidor y aumentando la actividad glutamatérgica excitatoria.
El resultado final sería un aumento de la excitabilidad neuronal que contrarrestaría los efectos depresores del alcohol. Al suprimir el consumo de alcohol, el efecto depresor desaparece pero, las neuronas permanecen hiperexcitables. Esta hiperexcitabilidad que requeriría tiempo para readaptarse, sería la responsable de la aparición de síntomas como la ansiedad, el insomnio y el craving.
***
El alcohol disminuye la actividad del receptor NMDA, lo que reduce la sensibilidad de las neuronas hipocampales y de las de Purkinje a la actuación del glutamato. La alteración del receptor podría justificar la pérdida de memoria producida por el etanol, así como la producción de convulsiones, que a veces acompañan a la abstinencia al alcohol. El tratamiento crónico con alcohol produce un aumento del número de receptores NMDA y de canales de calcio sensibles a voltaje, lo que podría aumentar la excitabilidad neuronal. También incrementa la transmisión GABAérgica. Como ya se indicó en el capítulo de los tranquilizantes, el alcohol actúa sobre este neurotransmisor mediante su unión al receptor GABA-A. Una confirmación de que el etanol afecta la neurotransmisión GABAérgica es que los alcohólicos crónicos tienen niveles reducidos de GABA en plasma y que al morir presentan en cerebro un número incrementado de receptores GABAérgicos.
Posteriormente, pueden aparecer convulsiones y/o delirium tremens. Se denomina delirium tremens a un estado de confusión agitada, asociado a veces a convulsiones o alucinaciones y/o desilusiones paranoides. Una posible explicación a la aparición de estos signos se basa en la disminución de la función de los receptores GABA-A tras la administración crónica de alcohol. La actividad reducida de las neuronas GABAérgicas puede disminuir la inhibición que el GABA ejerce sobre las neuronas noradrenérgicas y sobre los receptores NMDA, lo que aumenta la actividad de ambos. La elevación de los niveles de norepinefrina puede contribuir, a la disminución de los niveles tisulares de Mg++. Dado que estos iones inhiben los receptores NMDA, al disminuir su concentración aumentará la actividad de estos receptores.
La activación del receptor NMDA en áreas como e! hipocampo conduciría a la aparición de las convulsiones, así como en el hipocampo y otras regiones cerebrales a una liberación excesiva de dopamina. Esta difunción dopaminérgica puede contribuir a la aparición de las alucinaciones relacionadas con el delirium tremens.
El alcohol etílico también estimula la producción de serotonina e influye en los receptores de este neurotransmisor. El aumento en la producción de serotonina y el efecto estimulante del alcohol sobre el receptor 5-HT 3 incrementan la actividad neuronal al liberar impulsos eléctricos, lo que contribuye a estimular la liberación de otros neurotransmisores como la dopamina. Esto puede regular el consumo de alcohol al contribuir en los efectos de recompensa. Así mismo, la influencia del alcohol sobre el receptor 5-HT 2 puede contribuir a producir los síndromes de abstinencia. El exceso de serotonina también contribuye en los efectos tóxicos del alcohol.
Alcohol y el sistema opioide
Numerosas evidencias sugieren la existencia de un componente biológico en los mecanismos cerebrales de reforzamiento del alcohol. Las investigaciones en neurociencias se han centrado en el estudio de los sustratos neurales y los sistemas de neurotransmisores implicados en estos mecanismos. Varios estudios muestran que los sistemas dopaminérgico, serotoninérgico y de péptidos opioides en el cerebro juegan un papel clave en estos procesos. El alcohol aumenta la transmisión dopaminérgica y serotoninérgica en regiones cerebrales asociadas a las vías de recompensa. La administración de agonistas dopaminérgicos y serotoninérgicos reduce la ingesta de alcohol, mientras que la de antagonistas dopaminérgicos la aumenta. Algunos estudios sugieren que los receptores D2, 5-HT1A y 5-HT3 participan en estas respuestas. El alcohol y los péptidos opioides comparten muchas características farmacológicas y exhiben efectos similares sobre el comportamiento en animales y en el hombre. Se ha postulado al sistema opioide como posible mediador de los efectos reforzadores positivos del alcohol. El consumo de la sustancia es alterado por la administración de péptidos opioides exógenos, y el alcohol, a su vez, afecta la actividad del sistema opioide.
El etanol modifica la síntesis y la liberacíón de algunos péptidos opioides, así como la actividad de los receptores opiáceos mu y delta. Por otro lado, la administración de antagonistas selectivos de los receptores mu y delta reduce la preferencia por alcohol y la ingesta de la sustancia en animales. Los antagonistas opiáceos como la naltrexona, reducen las propiedades reforzadoras del alcohol en bebedores sociales y disminuyen la ingesta excesiva de la sustancia. En consecuencia, es posible que la preferencia por alcohol esté asociada con una activación aumentada del sistema opioide. El desarrollo de agentes farmacológicos capaces de modificar la transmisión de los péptidos opioides, así como la de otros neurotransmisores en el cerebro, tiene un uso terapeútico potencial para el tratamiento del alcoholismo en humanos.
Imagen de los receptores afectados por el alcoholEfectos Físicos del Alcohol:
- Muerte con una tasa del 0,50% de alcohol en sangre
- Muerte por asfixia, cierre de las vías respiratorias por obstrucción vómito etc.
- Debilidad agotamiento físico: disminución de azúcares, el alcohol acelera la transformación de glucógeno (una sustancia que se encarga de almacenar el azúcar en el hígado) en glucosa y ésta se elimina de forma más rápida.
- Inhibición de la vasopresina (hormona suprarrenal, control del balance de líquidos),cuya función es reabsorber el agua de la orina en los riñones, al faltar agua el organismo reabsorbe el agua de cualquier órgano, en el cerebro el alcohol reabsorbe el agua de las meninges produciendo cefaleas.
- Irritación gástrica (ardor de estómago) producido por el etanol erosionando la mucosa del estómago.
Fuentes:
- http://www.uvmnet.edu/investigacion/episteme/numero8y9-06/jovenes/a_etanol.asp
- http://bvs.insp.mx/articulos/5/1/102001.htm
- http://redalyc.uaemex.mx/pdf/582/58252401.pdf
- http://www.eurekalert.org/pub_releases_ml/2008-09/aaft-q082708.php
- http://www.biografica.info/redei/neurobiologia-de-la-drogradiccion
- http://www.analisisdedrogas.es/alcohol_26.html
Por razones más históricas que farmacológicas, se mencionaba este hongo como colinérgico, la Amanita muscaria. Decimos históricas porque se pensaba que los efectos tóxicos de este hongo se debían a sus interacciones con el receptor muscarínico de la acetilcolina (de ahí su nombre); sin embargo, se ha visto que no hay tal, y que sus efectos alucinógenos se deben más bien a sustancias que interactúan con el receptor del GABA, en este caso el muscimol, y del glutamato, mediante el ácido iboténico.
La amanita es uno de los hongos tóxicos más conocidos y fácil de reconocer: es el famoso hongo con sombrero rojo y puntos blancos de los cuentos. Se le ha asociado al Soma, la droga sagrada de la inmortalidad, mencionada en el Rig Veda (1500 a. C.), a la ambrosía de los dioses del Olimpo, a los misterios de Eleusis, también en la antigua Grecia, e incluso a los orígenes del cristianismo en Roma. Se sabe que lo usaban las tribus nómadas de Siberia, los chamanes, en particular en ritos comunales de indulgencia. Éstos conocían algo del metabolismo de los alcaloides responsables de los efectos alucinógenos del hongo y sabían que se excretaban en la orina, de manera que si ésta se bebía, se podían prolongar los efectos o compartirlos con un aprendiz. En las heladas regiones de Siberia, donde el hongo escasea, su precio era alto (en ocasiones, un solo hongo costaba un venado completo).
La intoxicación por muscarina provoca náusea, vómito, cefalea (dolor de cabeza), alteraciones visuales, arritmias cardiacas y estado de choque.
Fuente: http://omega.ilce.edu.mx:3000/__//html/sec_29.html
Pinchar imagen para agrandar.
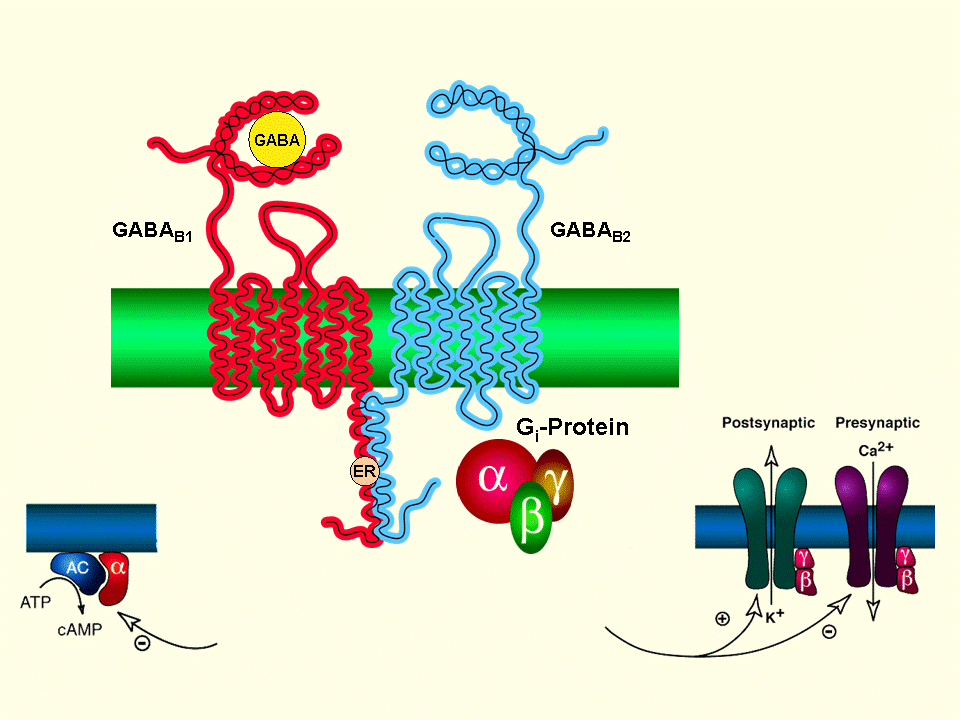
Receptor GABA-B:
Receptor metabotrópico del GABA-B unido a proteínas G y la adenilciclasa, lentamente inhibitorio, postsinápticos salida de K+ y presinápticos, cerrando los canales de Ca+, originando menor liberación de glutamato y de monoaminas.Receptor GBH:
Receptores acoplados a proteínas G donde se une el ácido gamma hidroxibutírico (GHB)
No Comparte ninguna homología de secuencia con el GABA B, causa un efecto estimulante seguido por convulsiones a dosis más altas, que se cree es mediado a través del aumento de Na +/K + y liberación de dopamina y glutamato.
Efectos La activación de ambos tipos de receptores, el específico del GHB y el del GABAB es responsable de sus efectos sedantes, somníferos y placenteros. Los efectos en el organismo dependerán de la dosis, ya que dependiendo de la concentración se activarán distintos receptores. Los efectos del GHB sobre la liberación de dopamina es bifásica,[30] concentraciones bajas de GHB estimulan el receptor específico estimulándose la liberación de dopamina.[31] Concentraciones más altas activan los receptores GABAB que inhiben la liberación de dopamina como hacen otras sustancias que sustituyen al GABA como el baclofen y phenibut.[32] Tras la fase inicial de inhibición, cuando se va metabolizando el GHB y disminuye la concentración, entonces la dopamina vuelve a liberarse al volver a activarse los receptores del GHB, regresando sus efectos estimulantes y placenteros. Tanto la inhibición como el incremento de la liberación de la dopamina se pueden bloquear con los antagonistas de los opioides tales como la naloxona y naltrexona. Esta paradójica mezcla de los efectos del GHB, sedante primero y estimulante después, denominado efecto rebote, es experimentado por los individuos consumidores de GHB como somnífero varias horas después de haber conseguido un sueño profundo inducido por el GHB. La aparición de esa baja concentración de GHB en el sistema provoca el estado de vigilia, por lo que suelen tener que tomar una segunda dosis en ese momento para permanecer dormidos.
Fuente: http://es.wikipedia.org/wiki/%C3%81cido_%CE%B3-hidroxibut%C3%ADrico
DROGAS OPIÁCEAS
(Analgésicas)
Alcaloides de núcleo fenantrénico
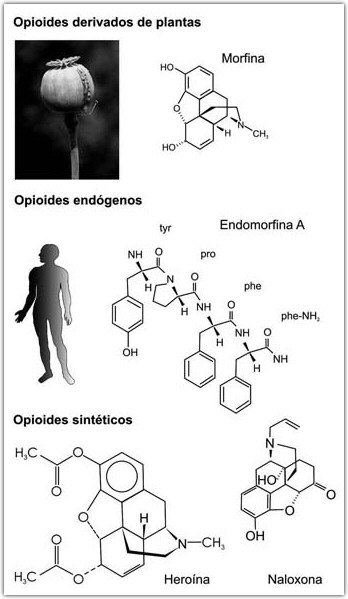

Drogas Opiáceas:
Receptor Mu (μ):Al igual que otras drogas de abuso, los opiáceos actúan sobre el sistema de recompensa. Como se indicó en el capitulo anterior, el sistema opioide endógeno también forma parte de dicho sistema, por lo que los opiáceos pueden mimetizar los mecanismos naturales de la recompensa. Los opiáceos actuan sobre la vía dopaminérgica mesolímbica en el área tegmental ventral, produciéndose una liberación de dopamina en el núcleo accumbens. En este área hay neuronas GABAérgicas con receptores mu cuya hiperpolarización disminuye la liberación de GABA sobre las neuronas dopaminérgicas, lo que contribuye a aumentar aún más la actividad de las células dopaminérgicas.
La dopamina podría estar implicada en los aspectos incentivos de la recompensa y, por tanto, en el mecanismo de reíorzamiento a la autoadministración de opiaceos. Una vez adquirido este hábito, su mantenimiento estaría relacionado no sólo con los aspectos incentivos, sino también con las propiedades consumatorias del estímulo opiaceo, que pueden ser independientes de dopamina. El comportamiento de búsqueda de droga es mantenido tanto por las propiedades reiorzadoras positivas, que producen una recompensa, como por las propiedades reforzadoras negativas, que son una consecuencia de la dependencia y que producen efectos aversivos. Estos últimos efectos podrían estar relacionados con la reducción de la actividad dopaminérgica que se produce en el sistema mesolímbico en la abstinencia. Esto indica un estado de dependencia a nivel de la respuesta de estas neuronas. La búsqueda de los opiáceos se le hace entonces necesaria al drogadicto para recuperar una neurotransmisión dopaminérgica normal y eliminar la disioria que acompaña a la abstinencia.
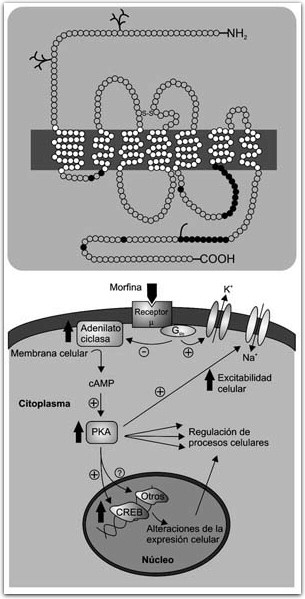
FIGURA 4. Estructura del receptor d; cada círculo representa un aminoácido. La barra negra representa la membrana celular. Los siete dominios transmembranales son típicos de receptores acoplados a proteínas G, con la terminal amino en la región extracelular y la carboxi en la intracelular. Los círculos obscuros representan sitios de fosforilación para proteínas cinasas A y C (PKA y PKC). Abajo, la activación de un receptor opioide (m) por la morfina, por ejemplo, produce una cascada de segundos mensajeros que inicia con la activación de una proteína G que puede modular directamente canales iónicos de la membrana celular o activar la adenilato ciclasa. Esto produce un aumento en los niveles de adenosin monofosfato cíclico (CAMP) lo cual a su vez modula la actividad de la proteína cinasa A (PKA). La PKA regula diversos procesos celulares, es capaz de modificar la actividad de canales iónicos y de regular la expresión genética uniéndose a la proteína CREB y a otros reguladores de expresión genética.
Receptor Kappa (κ):
En relación con el uso de las drogas es especialmente notable el hecho de que algunos agonistas de los receptores opioides k tienen un notable potencial alucinogénico. En particular, en años recientes se ha demostrado que la salvinorina-A, que es el principal producto activo de la Salvia divinorum (hojas de pastora o hierba de la virgen) es un potente agonista de los receptores k, y que es la activación de dichos receptores lo que explica el alto potencial alucinogénico de la Salvia divinorum.
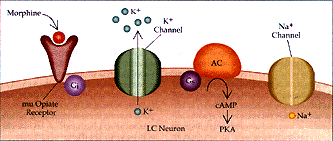
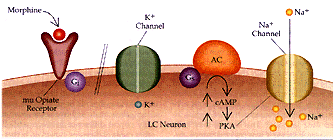
Figura 2 Sustrato neural para la dependencia física a opiáceos (a) Se muestra la acción de los opiáceos en las neuronas del locus cerúleo (LC) en un individuo no dependiente. La excitabilidad intrínseca de las neuronas del LC dependen de un canal de Na+ semejante al del marcapaso, que se activa por la vía del monofosfato cíclico de adenosina (AMPc). Los opiáceos actúan a través del receptor opiáceo mu y la proteína inhibitoria G, Gi, para inhibir la vía del AMPc y activar el canal inhibitorio de K+. (b) Con el uso prolongado de opiáceos, la cascada del AMPc se regula en forma positiva, incluyendo mayor producción de la proteina cinasa (PKA, o proteina cinasa A) dependiente de AMPc. La cinasa fosforila el canal de Na+ semejante al marcapaso, aumentando su eficacia. Además, el receptor opiáceo mu se desacopla de su conección con el canal inhibitorio K+ y no puede ya abrir el canal. Por estos mecanismos la administración de opiáceos a largo plazo puede aumentar la excitabilidad intrínseca de las neuronas del LC. AC= adenil ciclasa y Ge= proteína G estimuladora.
La base molecular de la tolerancia es de tipo farmacodinámico. Una de las teorías más reconocidas es la de la regulación por incremento del AMPc (up-regulation). Como se ha comentado, de forma aguda, los opioides disminuyen la concentración del AMPc y la actividad de la PKA. Tras la administración repetida, la actividad de la adenilciclasa y de la PKA se incrementan progresivamente (up-regulation) y, como consecuencia, van aumentando poco a poco las concentraciones de AMPc. Así, se necesitan cada vez dosis mayores de opioides para mantener la disminución de AMPc (tolerancia). Cuando deja de darse el opioide o se administra un antagonista como la naloxona, se produce un aumento de rebote del AMPc. Este gran incremento del AMPc aumenta la excitabilidad de las neuronas y es la base molecular de los signos y síntomas de la abstinencia. Este fenómeno se ha demostrado en el locus coeruleus, el núcleo accumbens, el área tegmental ventral y la sustancia gris periacueductal. Parece ser que, el causante de la regulación por incremento del sistema del AMPc es el aumento de la producción de un factor de transcripción llamado CREB (proteína que se une a elementos de respuesta a AMPc). Además, el CREB aumenta la síntesis de dinorfina, sustancia que activa los receptores kappa en las neuronas del área tegmental ventral, que produce una disminución de la liberación de dopamina en el núcleo accumbens. Esta reducción contribuye al estado emocional negativo (disforia y anhedonia) característico de la abstinencia. Otra teoría de la tolerancia postula que, la administración repetida de opioides produce una desensibilización de los receptores debido a su fosforilación por quinasas (quinasa del receptor de proteína G o GRK) y su unión a la β-arrestina, que pueden internalizarlo, con lo que disminuye la densidad de receptores en la membrana. La mayoría de agonistas provocan estas acciones rápidamente, mientras que, la morfina produce una internalización menor y más lenta. Por ello, no se conoce de forma exacta cuál es la base molecular de la tolerancia a la morfina.
Fuentes:
- http://www.biografica.info/redei/neurobiologia-de-la-drogradiccion
- http://www.analisisdedrogas.es/heroina_15.html
- http://www.drscope.com/privados/scientific/psiquiatria/ejemplo/index.html
- http://www.robertexto.com/archivo3/opioides.htm
- http://en.wikipedia.org/wiki/Opium
![]() Grados de abstinencia a las drogas opiáceas
Grados de abstinencia a las drogas opiáceas
La Heroína y sus adulterantes son inmunosupresores:
Immunoglobulin alterations associated with heroin addiction
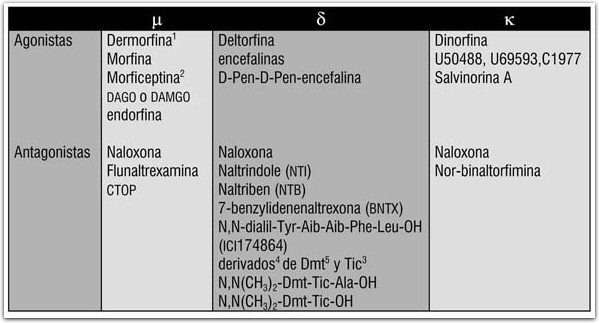
DROGAS CANABINOIDES
Inhibición del GABA y el Glutamato
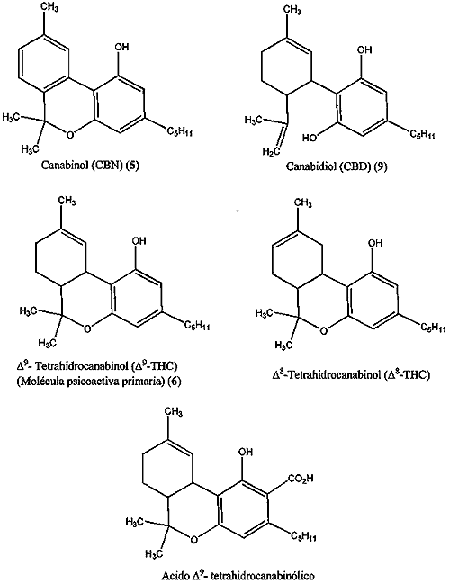
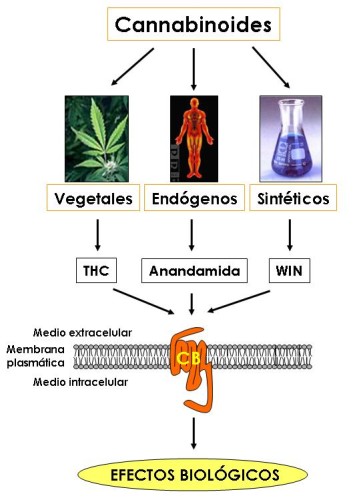

Drogas Canabinoides:
Luego de muchos años de investigaciones fue recién en 1964 que Rafael Mechoulam, de la Universidad Hebrea de Jerusalem, identificó al delta-9-tetrahidrocanabinol (THC), como el componente al que se atribuyen prácticamente todos los efectos medicinales de la marihuana.
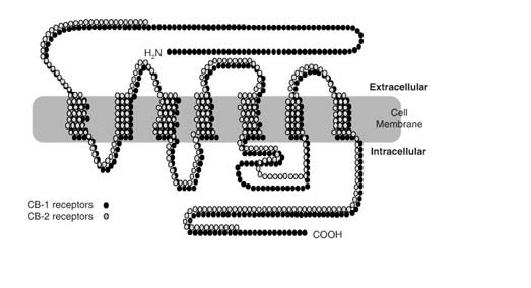
Ahora bien cómo es posible que una molécula de origen vegetal tenga efectos sobre el sistema nervioso central del hombre. La respuesta la proporcionó un laboratorio en EEUU, el cual demostró que el delta-9-THC se acopla a una proteína localizada en la membrana de las células. Este es un receptor que se denominó posteriormente como CB (canabinoides) y la nomenclatura de los receptores se amplió cuando se caracterizaron receptores en el SNC, los cuales fueron llamados CB1; mientras que aquellos que presentaban una distribución periférica en el sistema inmune eran referidos como CB2. Entonces los investigadores se preguntaron por qué una molécula producida por una
planta tenía receptores en el cerebro. Nuevamente el Dr. Mechoulam vino en nuestra ayuda descubriendo un pequeño ácido graso que era producido por el cerebro y que imitaba todas las actividades de la marihuana.
La llamó anandamida, (AEA) del sánscrito ananda, "el que trae bendición y tranquilidad interna".
En el año 1997 Daniele Piomelli y Nephi Stella, de la Universidad de California, descubrieron otro lípido, el 2-araquidonoil glicerol, (2-AG), aún más abundante que la anandamida, en ciertas regiones del cerebro. Ambos compuestos se consideran los principales canabinoides endógenos, o endocanabinoides.
Todos conocíamos a la marihuana, pero no todos sabíamos que nuestro cuerpo era capaz de fabricar su “propia marihuana”.
¿Neurotransmisores a pedido?
La mayoría de los neurotransmisores hasta ahora conocidos son solubles en agua y se almacenan en vesículas, a la espera de una señal para ser liberados por la neurona. Cuando una neurona "dispara", enviando una señal eléctrica por su axón hacia las terminales "presinápticas", los neurotransmisores liberados de sus vesículas cruzan la abertura sináptica y ejercen su efecto a través de sus receptores de la neurona "post- sináptica".
Sin embargo los endocanabinoides, son lípidos y no están almacenados en vesículas ni se encuentran pre-formados, sino que son rápidamente sintetizados a partir de sus precursores en la membrana celular, cuando los niveles de calcio dentro de la neurona se elevan o cuando se activan ciertas proteínas G.
Además, se había observado que los impulsos nerviosos se transmitían siempre en un sólo sentido, desde la célula presináptica hacia la postsináptica.
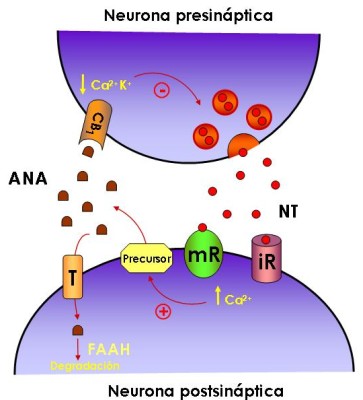
Figura 2: El sistema cannabinoide endógeno como sistema neuromodulador
NT, neurotransmisor; ANA, anandamida; T, transportador; FAAH, ácido graso amidohidrolasa; iR, receptor ionotrópico; mR, receptor metabotrópico; +, activación; -, inhibición. Para más detalles ver el texto.
Page 3 Revista QuímicaViva, número 3, año 4, diciembre 2005 88
Alger y sus colegas de la Universidad de Maryland y Marty de la Universidad de Paris observaron que tanto en las neuronas del hipocampo como del cerebelo las células receptoras parecían capaces de enviar a su vez señales "de vuelta" a las células emisoras. Esta señalización retrógrada es características de los endocanabinoides. Tanto la AEA como el 2-AG, son sintetizados en las células postsinápticas como las células piramidales del hipocampo. La síntesis se inicia por un influjo de calcio a través de canales dependientes de voltaje, o por la activación de una proteína G acoplada al receptor del neurotransmisor.
Los endocanabinoides llegan al espacio extracelular y activan los receptores CB1 que se encuentran en ciertas terminales nerviosas. La activación de los receptores CB1 conduce a la inhibición presináptica del GABA o a la liberación de glutamato al inhibir los canales de calcio y activar los canales de potasio. Los endocanabinoides pueden entrar a las células presinápticas o postsinápticas por el transportador de anandamida (AT). Además en las células postsinápticas está presente la FAAH (hidrolasa de amidas de ácidos grasos), enzima que degrada la anandamida y en las presinápticas la monoglicérido lipasa, que degrada el 2-AG.
“Ananda” te busco y no te encuentro…
"La cucaracha, La cucaracha
Ya no puede caminar, porque
le falta porque no tiene,
marihuana pa' fumar"
(Popular corrido de la revolución mexicana)
Fuente: http://www.quimicaviva.qb.fcen.uba.ar/V4n3/Franchi.html
Mecanismo de acción de los endocanabinoides
Se han realizado importantes descubrimientos sobre la función fisiológica de los endocanabinoides a partir de los estudios neurofisiológicos publicados independientemente por tres diferentes grupos de investigación en 2001. Se cree que actúan como mediadores sinápticos retrógrados del fenómeno de supresión de la inhibición (DSI) o de la excitación (DSE) provocada por la despolarización. El DSI es una forma de señal retrógrada rápida de las neuronas postsinápticas hacia las células inhibitorias que las inervan y ocurre predominantemente en el hipocampo o en el cerebelo. El DSE se produce principalmente en las células cereberales de Purkinje, como se evidenció en los estudios con ratas. Estos hallazgos sugieren que los endocanabinoides están involucrados en la modulación rápida de la transmisión sináptica en el SNC por el sistema de señales retrógradas, las cuales influyen sobre la sinapsis en un radio local de 40 µm de diámetro y causan efectos supresores sobre la liberación de neurotransmisores -tanto inhibitorios como excitatorios- que persisten por décimas de segundo. De este modo, parecen ejercer una función en el control de los circuitos neurales, especialmente en el cerebelo y el hipocampo. Los canabinoides exógenos no pueden remedar los efectos de los canabinoides endógenos debido a que causan una activación de larga duración de los receptores CB1 en todas las regiones cerebrales, de modo que su efecto global es producir una inhibición persistente de la liberación de neurotransmisores de las terminales nerviosas que expresan los receptores CB1 y, como consecuencia, ocluyen temporariamente y evitan los fenómenos DSI y DSE.
Efectos de los canabinoides sobre las funciones del SNC
Control psicomotor
Debido a que los receptores CB1 se expresan en altas densidades, principalmente en los ganglios basales y el cerebelo, los canabinoides ejercen efectos complejos sobre la función psicomotora. En efecto, los receptores CB1 se expresan en las neuronas gabaérgicas del cuerpo estriado y abundan en las regiones que contienen los axones terminales de estas células (globo pálido, núcleo endopeduncular y sustancia negra reticulada), así como en las terminales de las neuronas glutamatérgicas desde el núcleo subtalámico al globo pálido, núcleo endopeduncular y sustancia negra reticulada. Se cree que los canabinoides inhiben la liberación de GABA en el cuerpo estriado y de GABA y glutamato en los otros núcleos. Algunos autores sugirieron que la función principal del sistema endocanabinoide sería inhibir la liberación tónica del glutamato en la sustancia negra y regular los niveles de actividad motora basal. Los canabinoides exógenos pueden suprimir la liberación de GABA en la sustancia negra, lo que produce la desinhibición de las señales inhibitorias hacia la vía talamocortical, con la consiguiente inhibición del movimiento. Todavía no se ha aclarado cuánto de los efectos de los canabinoides sobre la función motora se deben a sus acciones sobre el cerebelo, aunque es probable que los ejercidos sobre la postura y el equilibrio se realicen en esta región, ya que hay gran cantidad de receptores CB1 próximos a los impulsos inhibitorios gabaérgicos o excitatorios glutamatérgicos hacia las células de Purkinje. Hay evidencia anecdótica acerca de que la cannabis puede aliviar el dolor muscular y la espasticidad en los pacientes con esclerosis múltiple.
Efectos sobre la memoria
Uno de los efectos mejor establecido de la intoxicación aguda con cannabis en el hombre es el deterioro en la memoria reciente. El sitio probable para estas acciones es el hipocampo, especialmente en las terminales de un subgrupo de interneuronas gabaérgicas que también contienen el neuropéptido colecistoquinina. También, los receptores CB1 se expresan en bajas concentraciones en las células piramidales glutamatérgicas. De esta manera, los canabinoides pueden inhibir la liberación de GABA y glutamato en los circuitos del hipocampo.
Efectos sobre la neocorteza
Al igual que en el hipocampo, la mayoría de las interneuronas corticales que expresan altos niveles de CB1 son gabaérgicas y contienen colecistoquinina. Los estudios de los efectos de los canabinoides sobre las habilidades perceptuales produjeron resultados disímiles. Mientras los consumidores a menudo comunican una mejoría subjetiva en la percepción visual y auditiva, los ensayos de laboratorio en general no demostraron cambios en la percepción. En cambio, un efecto subjetivo que sí se confirmó en los estudios de laboratorio es la sensación de que el tiempo pasa más rápido que el tiempo real. Aunque algunos ensayos demostraron signos leves de deterioro cognitivo en los consumidores crónicos de cannabis, hay poca evidencia de que sean irreversibles o se acompañen de neuropatología producida por drogas. Además, parecen asociarse con el consumo importante y a largo plazo y es poco probable que los experimenten aquellos que utilizan cannabis en forma recreativa.
Efectos hipotalámicos sobre el control del apetito
Ensayos clínicos controlados demostraron que el THC tiene efectos beneficiosos al contraponerse a la pérdida de apetito y la reducción de peso de los pacientes con sida con síndrome de impregnación; ésta es una de las indicaciones médicas por la cual la droga obtuvo la aprobación oficial en EE.UU. El THC también estimula la ingesta alimentaria en animales y el efecto es específico para alimentos dulces o con alto contenido graso. Estos resultados sugieren que la cannabis puede tener una función reguladora sobre la ingesta alimentaria y el peso corporal. Se postuló una relación recíproca entre los endocanabinoides hipotalámicos y la leptina, hormona supresora del apetito.
Efectos antieméticos y analgésicos
La capacidad del THC y del canabinoide sintético nabilona para controlar las náuseas y vómitos secundarios a quimioterapia constituye otra indicación médica para el uso de estas drogas. Sin embargo, la ventana terapéutica estrecha entre el efecto antiemético y las posibles reacciones adversas torna dificultosa su utilización; además, el advenimiento de los antagonistas de los receptores de 5HT3 más eficaces y seguros los hace menos atractivos.
La administración sistémica del THC y los canabinoides sintéticos produce efectos antinociceptivos y antihiperalgésicos en modelos animales de dolor agudo e inflamatorio. Evidencias experimentales con ratones sugirieron que no todos los efectos antinociceptivos son mediados por receptores CB1, sino que podría haber una interacción con los opioides. El THC y la morfina actuarían en forma sinérgica y su acción puede bloquearse por los antagonistas rimonabant o naloxona.
Los canabinoides como tóxicos y drogas de dependencia
A pesar de ser ilegal, la cannabis es una de las drogas más ampliamente utilizadas. La experiencia es altamente variable de acuerdo con la dosis, el ambiente y la expectativa del consumidor. La experiencia de plenitud es precedida por una etapa transitoria de sensaciones de estremecimiento u hormigueo en el cuerpo y la cabeza y vértigo. Se caracteriza por una rapidez de asociaciones mentales y sentido del humor, algunas veces descrita como euforia. Los usuarios en general se sienten relajados y calmos, en un estado de ensoñación, desconectados del mundo real. Los sujetos intoxicados a menudo demuestran dificultades en mantener una conversación coherente y pueden tener fantasías o soñar despiertos. También se refieren somnolencia y sensaciones de aumento de la percepción, del apetito y distorsión del sentido del tiempo. Los efectos adversos comunicados incluyen alteraciones en la memoria, paranoia y falta de motivación. Se conoce poco acerca del mecanismo cerebral involucrado en la sensación de euforia, aunque algunos creen que puede deberse a la interacción con los receptores CB1 del área límbica situados en las interneuronas gabaérgicas que contienen colecistoquinina. Otros autores lo atribuyen a la activación selectiva dopaminérgica del núcleo accumbens, en forma similar a la provocada por heroína, cocaína, anfetamina y nicotina. Los efectos tóxicos son claramente mediados por los receptores CB1.
Fuente: http://www.bago.com/BagoArg/Biblio/psiq181web.htm
Cannabis y Esquizofrenia
Dopamina
Se ha comprobado que la administración exógena de cannabinoides incrementa la actividad de las neuronas dopaminérgicas del área tegmental ventral o ATV, núcleo de origen de la vía mesolímbica (French y cols., 1997), lo que se asocia a un aumento de la liberación de dopamina en los centros diana mesolímbicos, principalmente a nivel del núcleo accumbens (Gardner y cols., 1988). Este hecho aparenta ser contradictorio porque a nivel sináptico la estimulación de los receptores CB 1 reduce la liberación de dopamina (Beltramo y cols., 2000; Wilson y Nicoll, 2002), pero la administración sistémica de agonistas CB 1 incrementa la liberación de dopamina en el núcleo accumbens porque en el ATV los receptores CB 1 están en neuronas inhibitorias GABA y ocasionan un descenso de la inhibición GABA sobre las neuronas de dopamina, dando lugar a un incremento neto de la actividad dopaminérgica en esta región cerebral
Glutamato
. En lo que respecta al sistema endocannabinoide, el 2-AG (y en mucha menor medida la AEA que actúa más sobre la liberación de dopamina y GABA) modula la liberación de glutamato actuando sobre receptores CB 1 presinápticos, reduciéndola (Katona y cols., 2006). Se postula que, de modo semejante a como la hacen otros agonistas cannabinoides, el 2-AG reduciría la liberación de glutamato en regiones cerebrales involucradas en la esquizofrenia como el hipocampo (Fujiwara y Egashira, 2004), la corteza prefrontal (Auclair y cols., 2000), el núcleo acumbens (Robbe y cols., 2001) y la amígdala (Azad y cols., 2003). Además, la hipoactividad glutamatérgica en la corteza prefrontal podría influir sobre la hipofunción dopaminérgica detectada en esta estructura en la esquizofrenia, pues una menor liberación de glutamato prefrontal se acompaña de una menor liberación de dopamina, mediada por los complejos receptoriales NMDA/D 1 de las terminales de dopamina prefrontales (véase figura 1.2). El 2-AG actúa sobre receptores CB 1 , lo que explica que el SR141716A anule los efectos disruptivos del filtrado sensorial de los antagonistas NMDA o los efectos “psicóticos” del delta-9-THC agudo en modelos animales (Fujiwara y Egashira, 2004). A nivel estriatal se sabe que la liberación de glutamato estimula la producción de 2-AG a través de receptores mGLUR5 postsinápticos, lo que a su vez suprime la liberación de glutamato a través de un mecanismo retroactivo (Katona y cols., 2006). El 2-AG jugaría por tanto un papel fisiológico distinto a la anandamida en los trastornos psicóticos, y siempre conviene recordar que la concentración de 2-AG es 1000 veces superior a la de AEA en el cerebro (Bortolato y cols., 2006).
DROGAS XANTINAS
(Estimulantes suaves)
Xantinas Metiladas
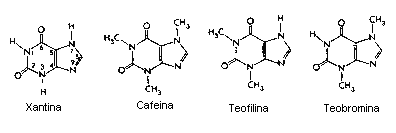
Drogas Xantinas:
De acuerdo a los expertos, la cafeína, que pertenece al grupo de sustancias llamadas xantinas, estimula el cerebro al interferir en la acción de la adenosina un transmisor nervioso que produce calma y tranquilidad. Los efectos farmacológicos de la adenosina pueden ser minimizados en individuos que esten tomando grandes cantidades de estimulantes tipo Metilxantina, por ejemplo la ya mencionada teofilina (presente en el té), la teobromina (en el chocolate) y la cafeína (en el café). Los efectos estimuladores del café son principalmente (aunque no enteramente) acreditados a su capacidad de inhibir la adenosina al competir por los mismos receptores, por razón del componente de purina en la estructura de la cafeína, bloqueando eficazmente los receptores de la adenosina en el SNC. Esta reducción de la actividad de la adenosina conlleva a una incrementada actividad de neurotransmisores como la dopamina y el glutamato provocando una sensación de euforia y de fuerza durante algunas horas. También facilita la actividad intelectual y la creatividad, al mantener despierto y en estado de alerta a su consumidor. Todo esto ocurre junto con un incremento de los niveles de adrenalina y noradrenalina, que son neurotransmisores activadores.
“La cafeína inhibe la fosfodiesterasa, que es responsable de la desactivación del AMPc. El crecimiento de la tasa de AMPc intra-celular, amplifica sus acciones de « segundo mensajero », lo que lo hace responsable de las principales consecuencias farmacológicas de la cafeína.” (Katzung, 1991)
La máxima concentración en la sangre se alcanza entre los 30 y 45 minutos de haberla ingerido. A las tres horas ya se ha eliminado la mitad de lo que se absorbió.
Según el libro de Farmacología, del doctor Manuel Litter, esta sustancia actúa en distintos niveles en todo el cuerpo. A dosis habituales de dos a cuatro tazas diarias -150 a 250 miligramos- estimula las funciones psíquicas, lo que aparentemente no es seguido de depresión; y se hace más fácil el esfuerzo intelectual, la asociación de ideas y la atención. En el nivel del sistema cardiovascular actúa estimulando el corazón -incrementa la frecuencia cardíaca y la fuerza de contracción- y además aumenta la presión arterial en forma transitoria.
Tanto la cafeína como la teofilina provocan disminución del flujo sanguíneo cerebral por vasoconstricción, aliviando de esta manera la cefalea.
Otro efecto importante es que aumenta la secreción de jugos -como el ácido clorhídrico y la pepsina- en el estómago. Esta acción la convierte en una droga irritante de la mucosa gástrica; pero, a su vez, tiene acción antiespasmódica en la vesícula.
La cafeína posee también un leve efecto diurético; aumenta la capacidad de trabajo muscular, refuerza la contracción, retarda y alivia la fatiga. Finalmente, produce un muy pequeño efecto en los pulmones, dilatando los bronquios.
Fuentes: http://www.buenasalud.com
http://www.infarmate.org/pdfs/noviembre_diciembre/cafeina.pdf
SOLVENTES E INHALANTES

Solventes e Inhalantes:
El principal problema de abuso de drogas es el de los inhalantes. Se encuentra en el ámbito laboral, lo cual no es nada trivial, pues implica a miles de trabajadores de la industria petrolera, la química, las imprentas, las gasolineras, a los pintores, carpinteros, zapateros, etc., pero de manera preocupante, es un problema que adquiere características dramáticas en la población más afectada, la de niños y jóvenes, la vasta mayoría, de clases desfavorecidas. Independientemente del país o continente donde se haya realizado el estudio, las conclusiones apuntan siempre hacia la misma dirección: el origen más probable del inhalador es de las clases marginadas, aquellas donde la pobreza, la falta de educación y de oportunidades es endémica, allí donde las familias se encuentran desmembradas o inexistentes, allí donde la solidaridad es más bien complicidad con otros que corren la misma suerte.
El término inhalantes se refiere al grupo de sustancias psicoactivas que se definen más por su modo de administración que por su mecanismo de acción o farmacología. Excluyendo a otras sustancias que también se inhalan como el tabaco, la mariguana, el opio o la cocaína, aquí se incluye más bien a un grupo de sustancias volátiles (esto quiere decir que su punto de ebullición es bajo, o en otras palabras, "hierven" a temperatura ambiente) que se utilizan para alterar el estado mental y que rara vez se administran por otra vía que no sea la inhalación. De acuerdo con esta definición se han establecido varias categorías de sustancias:
a) Gases anestésicos de uso médico: éter, cloroformo, halotano, óxido nitroso; (TAMBIÉN DISOCIATIVOS)
Farmacología del Sistema Nervioso
b) Solventes industriales o domésticos, incluyendo los adelgazadores (thinners) de pintura o solventes, los desengrasadores y los solventes de los pegamentos;
c) Los solventes contenidos en artículos de papelería o de arte, como los líquidos correctores o los solventes de los plumones;
d) Gases usados en la casa o la industria, como el gas para encendedores, los sprays de crema batida o los usados para limpiar circuitos electrónicos o los gases para rellenar refrigeradores;
e) Los aerosoles domésticos para aplicar pintura, fijadores para el cabello, protectores de tela, etc., y
f) Los nitritos alifáticos (medicamentos vasodilatadores).
El efecto agudo de la intoxicación con solventes es semejante a una borrachera: el sujeto muestra una excitación inicial que se convierte en desinhibición, con una sensación de ligereza, euforia y agitación. Cuando la dosis aumenta se puede observar ataxia, disminución de los reflejos, mareo y desorientación. En casos de intoxicación severa se produce debilidad muscular, alteraciones del lenguaje, nistagmus (los ojos oscilan en forma involuntaria), delirio y ocasionalmente alucinaciones con conductas francamente alteradas. Algunas horas después, el sujeto puede despertarse con una especie de "cruda": dolor de cabeza, letargo, incoordinación muscular, desorientación, etcétera.
El cuadro dependerá del solvente en particular, aunque frecuentemente los sujetos se administran mezclas de ellos, que hace casi imposible adjudicar la intoxicación a un solo agente. Esta interacción de solventes es significativa, no sólo porque la mezcla puede dar efectos mayores que los producidos por las sustancias aisladas, sino también porque pueden producirse metabolitos activos o cuya presencia potencie a otros. Por ejemplo, el hexano produce dos metabolitos: la metilbutilcetona (MBC) y la metiletilcetona.(MEC). Sola esta última no es tóxica, pero combinada con la MBC o el hexano mismo, potencia el efecto de aquéllos.
A pesar de que los inhalantes no produzcan tolerancia o dependencia física, sí causan una amplia variedad de efectos tóxicos. En general, y a nivel neurológico, éstos son de carácter difuso en sus manifestaciones, no muestran alteraciones focales, de manera que pueden confundirse con trastornos metabólicos, degenerativos, nutricionales o desmielinizantes, que también son difusos. Además, el daño es difícil de detectar, aún con las técnicas modernas de imagenología (véase el capítulo 1), como la tomografía computarizada, estudios electrofisiológicos de conducción nerviosa o resonancia magnética nuclear.
Los efectos asociados, sea a la intoxicación aguda severa o a la crónica leve con solventes orgánicos, son en general reversibles. Los daños graves e irreversibles ocurren usualmente en casos de intoxicación severa (concentraciones varios cientos de veces más altas que las existentes en un medio laboral) que se da durante largo tiempo, condiciones que sólo se presentan en el contexto del abuso de drogas. Algunas de las manifestaciones del daño neurológico pueden ser parcialmente reversibles cuando se suspende la inhalación y éstas concluyen al interrumpir la administración. Es decir, si el sujeto deja de inhalar, la toxicidad se interrumpe y no avanza más.
Los principales síndromes neurológicos producidos por los solventes orgánicos son los siguientes:
a) Encefalopatía: aguda o crónica, dependiendo del nivel y tiempo del consumo;
b) Ataxia cerebelosa: manifestada básicamente por trastornos del equilibrio y de los movimientos oculares;
c) Neuropatía periférica: los nervios de las extremidades degeneran a partir de la periferia, en dirección del centro (axonopatía distal); se observa pérdida de la sensibilidad, sin dolor;
d) Neuropatía craneal: con afectación de los nervios trigémino y facial;
e) Parkinsonismo;
f) Pérdida de visión (neuropatía óptica);
g) Alteraciones multifocales: demencia, ataxia, espasticidad, disfunción de estructuras del tallo cerebral, etcétera.
La gravedad de estas alteraciones dependerá, como ya dijimos, de la intensidad del abuso, es decir: I) el tiempo que se lleva inhalando, 2) el o los solventes utilizados, y 3) la dosis (frecuencia y cantidad) del inhalante.
Fuente: http://omega.ilce.edu.mx:3000/__/130/html/sec_36.html
Anatomía Núcleo Accumbens
El núcleo accumbens es un grupo de neuronas del encéfalo, localizadas en el lugar donde el núcleo caudado y la porción anterior del putamen confluyen lateralmente dispuestos con respecto al septum pellucidum. El núcleo accumbens se puede dividir en dos estructuras, la zona central y la corteza.
Estas estructuras tienen diferente morfología y función. El núcleo accumbens y el bulbo olfatorio forman colectivamente la parte ventral del cuerpo estriado, que es parte de los ganglios basales. Se piensa que este núcleo tiene un papel importante en la recompensa, la risa, el placer, la adicción y el miedo.
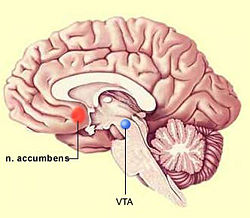
El principal tipo de neuronas que se encuentra en el núcleo accumbens es la neurona de proyección espinosa media. El neurotransmisor producido por esas neuronas es el GABA, uno de los principales neurotransmisores inhibidores del sistema nervioso central.
Estas neuronas también son la mayor proyección u output del núcleus accumbens. Aunque éste último tipo celular constituye el 95% de los efectivos en éste núcleo, también se pueden encontrar otras como las interneuronas colinérgicas grandes no espinosas. Aferencias y eferencias.
Las neuronas eferentes del núcleo accumbens proyectan sus axones hacia sus análogas de la parte ventral del globus pallidus (pálido ventral o VP). Éste a su vez proyecta hacia el núcleo medio dorsal del núcleo dorsal del Tálamo, que proyecta hacia la corteza prefrontal. Entre otras eferencias del nucleo accumbens se incluye las conexiones con la substancia nigra y la formación pontina reticular.
Las principales aferencias del núcleo accumbens son las cortezas prefrontales asociativas, la amígdala y las neuronas dopaminérgicas localizadas en el área ventral tegmental, que se conecta a través de la vía mesolímbica. Así pues se describe frecuentemente al núcleo accumbens como parte del bucle cortico-estriado-tálamo-cortical. Se piensa que los impulsos dopaminérgicos del área ventral tegmental modulan la actividad de las neuronas del núcleo accumbens. Estas terminales dopaminérgicas provenientes del área ventral tegmental son el sitio de acción de drogas altamente adictivas como la cocaína y la anfetamina, las cuales provocan un aumento en la liberación de dopamina en el núcleo accumbens.
Además de la cocaína y la anfetamina, se ha visto que casi todas las drogas de uso recreativo (heroína, morfina, nicotina) son capaces de incrementar, por diversos mecanismos, los niveles de dopamina en el núcleo accumbens. Trabajos de investigación En los años 50 Olds y Milner implantaron electrodos en el área septal de las ratas y encontraron que éstas elegían presionar la palanca que lo estimulaba. Esta preferencia era superior a la necesidad de comer o beber. Esto sugería que esta área es el "centro del placer" del cerebro. Aunque se ha estudiado tradicionalmente el nucleus accumbens por su papel en la adicción, desempeña un papel similar en recompensas como la de la alimentación, el sexo y los videojuegos. Un estudio reciente encontró que está implicado en las emociones inducidas por la música , quiza en consecuencia con su papel mediador de la liberación de la dopamina. También interviene en las pautas temporales, y se ha considerado ampliamente como la interfaz entre el sistema límbico y el motor (Mogensen).
Fuente: http://es.wikipedia.org/wiki/N%C3%BAcleo_accumbens
Comportamiento compulsivo
Estudios con animales de laboratorio, iniciados hace ya unos cuarenta años, han permitido comprobar que varias drogas siguen una misma trayectoria cerebral en su progreso del consumo a la adicción. Si se les brinda la oportunidad, ratas, ratones y primates no humanos se autoadministrarán las mismas substancias con las que se droga el hombre. En estos experimentos, los animales se conectan a una vía de inyección intravenosa. Aprenden a apretar tres palancas: una para recibir una infusión intravenosa de droga, otra para inyectarse una solución salina inocua y una tercera para alcanzar el alimento. En pocos días, los animales se han habituado: se autoadministran con toda facilidad cocaína, heroína. anfetamina, nicotina y otras. Aún más, acaban presentando comportamientos típicos de la adicción. Para drogarse, son capaces de renunciar a la comida y el sueño; algunos incluso llegan a morir por agotamiento o malnutrición. En el caso de las sustancias más adictivas, como la cocaína, los animales pasarán la mayor parte del tiempo en vigilia, afanándose por conseguir otra dosis, aun cuando eso signifique tener que presionar la palanca cientos de veces para obtenerla. Igual que los adictos humanos, a los que cualquier referencia a la droga (sea el lugar de consumo o los útiles de su aplicación) les provoca una intensa ansiedad por conseguir una dosis, los animales terminan por preferir un entorno relacionado con el estimulante (la zona de la jaula donde se halla la palanca que les proporciona bienestar). Cuando se les quita la sustancia, los animales pronto abandonan ese afán por la compensación química. Pero no olvidan el placer. Una rata que haya permanecido "limpia", incluso durante meses, volverá a presionar de forma compulsiva la palanca si se le deja probar cocaína o se le encierra en una jaula que ella asocia con su consumo. De modo parecido. las señales que puedan convertirse en indicadores del suministro de droga, como una descarga eléctrica en la pata, provocarán que la rata vuelva a la droga. Los mismos tipos de estímulos, exposición a dosis bajas o signos vinculados al estupefaciente despiertan el deseo y provocan la recaída en el adicto humano. Apoyándose en tales experimentos de autoadministración, se han cartografiado las regiones del cerebro que participan en el comportamiento adictivo y se ha descubierto la función principal del sistema de recompensa. Las drogas se hacen con el control del sistema y estimulan su actividad con una energía y persistencia superiores a cualquier recompensa natural. Un componente clave del circuito de recompensa es el sistema mesolímbico de la dopamina: un conjunto de células nerviosas que tienen su origen en el área tegmental ventral (ATV, cerca de la base del cerebro) y dirigen sus proyecciones hacia determinadas regiones del prosencéfalo, muy en particular hacia el nucleus accumbens. Esas neuronas del ATV se comunican enviando dopamina (un neurotransmisor) desde los extremos de sus axones hasta los receptores alojados en las neuronas del nucleus accumbens. La vía de la dopamina desde el ATV hasta el nucleus accumbens resulta decisiva para la adicción: los animales que sufren una lesión de esas regiones no vuelven a mostrar interés alguno por las sustancias adictivas.
Evaluación de la recompensa
Las vías de la recompensa poseen una larga historia evolutiva. El propio Caenorhabditis elegans, un gusano muy elemental, presenta una versión, rudimentaria, del sistema. En estos organismos, la inactivación de las neuronas dopaminérgicas clave, entre cuatro y ocho, provoca que pasen de largo, sin prestarle la menor atención, ante un tapiz de bacterias, su alimento favorito.
En los mamíferos, el circuito de recompensa reviste mayor complejidad. Opera con otras regiones del cerebro para conferir emotividad a una experiencia y dirigir la respuesta del individuo ante los estímulos de recompensa, incluidos el alimento, el sexo y la interacción social. La amígdala, por ejemplo, ayuda a valorar si una vivencia resulta agradable o repulsiva, si debería repetirse o evitarse; contribuye también a entablar conexiones entre una y otros indicios de referencia. El hipocampo participa en el registro de recuerdos de un hecho: dónde, cuándo y con quién ocurrió. Por fin, las regiones frontales de la corteza cerebral coordinan y procesan toda esta información y determinan el comportamiento ulterior del individuo.
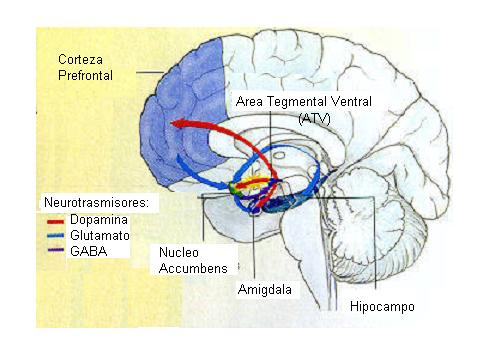
La vía ATV-accumbens, mientras tanto, "indica" a los otros centros del cerebro cuán satisfactoria es una actividad. Cuanto más nos satisfaga, tanto más probable es que el organismo la recuerde e intente repetirla.
Aunque buena parte de nuestro conocimiento sobre el sistema cerebral de recompensa lo hemos adquirido a través de modelos animales, la investigación llevada a cabo en el último decenio mediante técnicas de formación de imágenes ha corroborado en el hombre la existencia de vías equivalentes relativas al control de la recompensa natural y de la mediada por drogas. El recurso de la resonancia magnética funcional o tomografía de emisión de positrones (técnicas que miden cambios de flujo sanguíneo relacionados con la actividad neuronal) nos ha permitido observar la excitación del nucleus accumbens de un cocainómano cuando se le ofrece una "raya".
Si a ese mismo drogadicto se le muestra un vídeo en que aparece una persona consumiendo cocaína o una fotografía de rayas blancas sobre un espejo, su nucleus accumbens responde de forma semejante, junto con la amígdala y ciertas áreas de la corteza. Las mismas regiones se excitan en los ludópatas cuando contemplan imágenes de máquinas tragamonedas, lo que sugiere que la vía ATV-accumbens desempeña también un papel crítico en adicciones no relacionadas con las drogas.
Fuente: http://www.dejardefumar.com.ar/articulos.asp?art=78&cat=4
Drogodependencia
http://psicofarmacologia.info/drogas.html

Pinchar ver en grande
La heroína modifica la acción de la dopamina en el núcleo accumbens y el área tegmental ventral del cerebro - estas áreas forman parte de "circuito de recompensa" del cerebro. Una vez que cruzan la barrera hematoencefálica, la heroína se convierte en morfina, que actúa como un agonista de gran alcance en el subtipo de receptores opioides mu. Esta unión inhibe la liberación de GABA de la terminal nerviosa, lo que reduce el efecto inhibitorio de GABA en las neuronas dopaminérgicas. La mayor activación de las neuronas dopaminérgicas y la liberación de dopamina en la sinapsis y la activación sostenida de la membrana post-sináptica;produce la activación continua de la vía de recompensa dopaminérgica conduce a los sentimientos de euforia y el sentimiento de bienestar asociadas con el consumo de heroína. La morfina es un agonista débil en la opioides kappa y delta subtipos del receptor y la activación de estos receptores tiene un débil efecto en la activación de la vía de recompensa dopaminérgico.
Fuente: http://www.cnsforum.com/content/pictures/imagebank/hirespng/moa_heroin_mu.png
Para ver más ilustraciones de la actuación de las drogas:
http://www.cnsforum.com/imagebank/section/substance_abuse/default.aspx
Dependencia
Escala racional para evaluar el daño de las drogas (media daño físico y la media de dependencia).
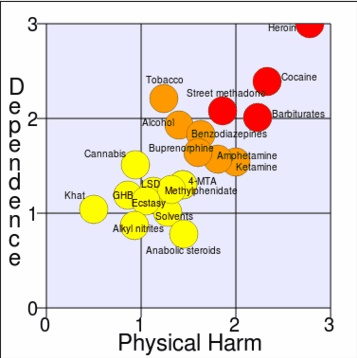
Genética
Alelo D2R2
El descubrimiento reciente del alelo D2 R2 por los investigadores Blum y Cumings ( un alelo es una variante de un gen normal ) el cual se encuentra en el 50 al 80 % de los alcohólicos, drogadictos, comilones y jugadores compulsivos hace renacer las esperanzas sobre un tratamiento biológico común para las personas que padecen estas enfermedades.El gen D2 R2 impide que la dopamina se acople a las células en la vía de recompensa, de manera que se reduce la corriente de placer que normalmente desata este neurotransmisor al liberarse, por tanto la gente portadora del gen mencionado tendría la necesidad de consumir ó hacer mas y mas cosas de lo que en general se satisfacen mediante la liberación de dopamina.
Según Blum y Cumings, el "hambre de dopamina" seria la responsable de generar los tipos ya descritos de comportamientos neuróticos y autodestructivos y las esperanzas surgen de la posibilidad de generar tratamiento de ingeniería genética molecular que permita la corrección de dicho defecto ó del desarrollo de un fármaco que neutralice la actividades alelo sobre el acoplamiento de la dopamina y que actué selectivamente sobre el sistema de recompensa.
Fuente:
- http://www.monografias.com/trabajos10/buspi/buspi.shtml
- Dopamina y Aprendizaje
- http://www.maikelnai.es/2008/01/22/los-hay-que-nunca-aprenden/
- Patrones y Dopamina
- http://www.microsiervos.com/archivo/azar/patrones-dopamina.html
TODO sobre Drogas y Drogodependencia
- http://wapedia.mobi/en/Psychoactive_drug
- http://psicofarmacologia.info/drogas.html
- http://www.erowid.org/splash.php
- http://www.cannabiscafe.net/
- http://www.psiconautica.org/
- http://www.mind-surf.net/drogas/
- http://www.cat-barcelona.com/ret/pdfret/RET42-1.pdf
- http://www.slideshare.net/drbobe/16-t/


6 meses para responder posts antiguos
¡Queremos tu opinión!
Si pregunta sobre este fármaco revise antes estos dos enlaces en particular
Ahora ya puede entender el prospecto y la ficha técnica

