

Búsqueda Simple
Búsqueda Completa
Taxonómica
Anatomía

+ Teoría
Patología
Aleatorio Principio Activo/ComercialFármacos o Drogas Más Visitadas
Levomepromazina

708715 visitas
Desvenlafaxina

274646 visitas
Clobenzorex
273269 visitas
Quetiapina

242386 visitas
Acepromazina

209803 visitas
Últimos comentarios


Sigue las noticias por


Total Fármacos Visitados
15.475.292
Desde Noviembre de 2008
CLASIFICACIÓN VARIOS


Nombre: Gabapentina
Comercial: Neurontin
Foto: 
Fórmula: 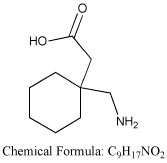
http://www.eutimia.com/psicofarmacos/anticiclicos/gabapentin.htm
Fármaco consultado: 50161 veces
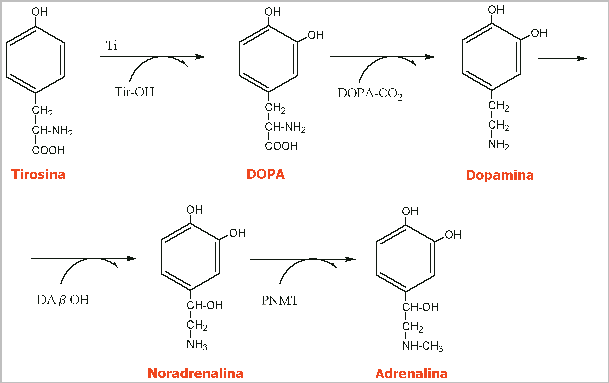

La adrenalina, también llamada epinefrina, es una hormona vasoactiva secretada en situaciones de alerta por las glándulas suprarrenales. Es una monoamina catecolamina, simpaticomimética derivada de los aminoácidos fenilalanina y tirosina. A veces es llamada "epi" en la práctica médica.
La adrenalina (en latín'ad' significa ‘al lado’ y «renal» viene de ‘riñon’) debería llamarse «suprarrenalina» ya que las glándulas suprarrenales se encuentran ‘encima’ (supra) de los riñones. El nombre epinefrina, (en griego "epi" significa "arriba" y "nefron" es riñón), Se diferencia de la noradrenalina, o norepinefrina, en que su efecto es más rápido y corto. Aunque se nombra de manera general como adrenalina fuera de los Estados Unidos
Efectos
Ante todo, la adrenalina es una hormona de acción, secretada por las glándulas adrenales en respuesta a una situación de peligro. Su acción está mediada por receptores adrenérgicos, tanto de tipo a como ß. Entre los efectos fisiológicos que produce están:
- Aumentar, a través de su acción en hígado y músculos, la concentración de glucosa en sangre. Esto se produce porque, al igual que el glucagón, la adrenalina moviliza las reservas de glucógeno hepático y, a diferencia del glucagón, también las musculares.
- Aumentar la tensión arterial: esto se produce en las arteriolas, en las que tiene lugar una vasoconstricción que provoca un aumento de la presión.
- Aumentar el ritmo cardíaco.
- Dilata la pupila para tener una mejor visión.
- Aumenta la respiración, por lo que se ha usado como medicamento contra el asma.
- Puede estimular al cerebro para que produzca dopamina, hormona responsable de la sensación de bienestar, pudiendo crear adicción.
La adrenalina y los compuestos relacionados producen efectos adrenérgicos que son tanto excitadores como inhibidores. Aquellas respuestas atribuidas a la activación de un receptor alfa son primariamente excitadoras, con la excepción de la relajación intestinal. Aquellas respuestas atribuidas a la activación de un receptor beta son primariamente inhibidoras, con la excepción de los efectos estimulantes miocárdicos.
| Receptor Alfa | Receptor Beta |
| Vasoconstricción (cutánea, renal, etc.) | Vasodilatación (músculo esquelético, etc.) |
| Contracción de la cápsula esplénica | Cardioaceleración |
| Contracción del miometrio | Aumento de la fuerza de contracción del miocardio |
| Contracción del dilatador del iris | Relajación del miometrio |
| Contracción de la membrana nictitante | Relajación bronquial |
| Relajación intestinal | Relajación intestinal |
| Contracción pilomotora | Glucogenólisis |
| Lipólisis | Calorigénesis |
La adrenalina es el activador más potente de los receptores alfa, es 2 a 10 veces más activa que la noradrenalina y más de 100 veces más potente que el isoproterenol. La adrenalina y los compuestos relacionados producen efectos adrenérgicos que son tanto excitadores como inhibidores. Aquellas respuestas atribuidas a la activación de un receptor alfa son primariamente excitadoras, con la excepción de la relajación intestinal. Aquellas respuestas atribuidas a la activación de un receptor beta son primariamente inhibidoras, con la excepción de los efectos estimulantes miocárdicos. Bajo la influencia de la adrenalina, la sístole ventricular se vuelve más rápida y de mayor fuerza, la duración de la sístole se acorta y la relajación diastólica se hace más rápida. Este tipo de acción inotrópica es independiente de la frecuencia cardiaca y es un efecto adrenérgico específico.
http://es.wikipedia.org/wiki/Adrenalina

La noradrenalina norepinefrina actúa como la noradrenalina, pero es sintética) es un neurotransmisor de catecolamina de la misma familia que la dopamina y cuya fórmula estructural es C8H11NO3.
Los cuerpos celulares que contienen noradrenalina están ubicados en la protuberancia y la médula, y proyectan neuronas hacia el hipotálamo, el tálamo, el sistema límbico y la corteza cerebral. Estas neuronas son especialmente importantes para controlar los patrones del sueño. Se demostró que la eliminación de noradrenalina del cerebro produce una disminución del impulso y la motivación, y se puede relacionar con la depresión. Además tiene que ver con los impulsos de ira y placer sexual.
La noradrenalina a su vez, funciona como neurotransmisor (junto con la adrenalina) de las vías simpáticas del Sistema Nervioso Autónomo, en las sinapsis postganglionares, que inervan los órganos blanco. Los receptores para la noradrenalina en las membranas postsinápticas de estas sinapsis son los receptores del tipo alfa y tipo beta. Generalmente, dichos receptores son antagonistas.
En la transmisión sináptica de este neurotransmisor, la síntesis está precursada por una enzima llamada dopaminabetahidroxilasa (DA-ß-hidroxilasa). Su liberación depende de la liberación de calcio, y teniendo dos tipos de receptores, los metabotrópicos y los iónicos, aunque los más importantes sean los primeros: ß1 (Beta1) respuesta postsináptica; ß2 (Beta2) respuesta postsináptica; a1 (Alfa1) hiperpolarización y a2 (Alfa2) autorreceptores.
Actúa sobre las células efectoras al unirse a unos receptores específicos, que pueden ser de dos tipos: receptores adrenérgicos alfa o receptores beta. Los receptores alfa intervienen en la relajación intestinal, la vasoconstricción y la dilatación de las pupilas. Los receptores beta participan en el aumento de la frecuencia y contractilidad cardiacas, la vasodilatación, la broncodilatación y la lipolisis.
Su destrucción lleva a cabo mediante recaudación, donde se ven implicadas las enzimas MAO (monoamino oxidasa) y COMT. El principal núcleo de producción de Noradrenalina es el Locus Coeruleus.
Un alto nivel de secreción de Noradrenalina aumenta el estado de vigilia, incrementando el estado de alerta en el sujeto, así como también facilita la disponibilidad para actuar frente a un estímulo. Y, contrariamente, unos bajos niveles de ésta secreción causan un aumento en el sueño y, también, estos bajos niveles pueden ser una causa de la depresión.
- http://es.wikipedia.org/wiki/Noradrenalina
- http://canal-h.net/webs/sgonzalez002/Farmaco/ADREN%C9RGICA.htm
Receptor adrenérgico
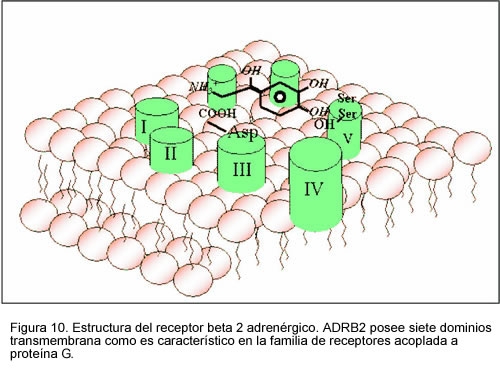
Los receptores adrenérgicos o adrenoreceptores son una clase de receptores asociados a la proteína G, los cuales son activados por las catecolaminas adrenalina (epinefrina) y noradrenalina (norepinefrina). Existen muchas células que poseen estos receptores y, la unión de un agonista adrenérgico causará, por lo general, una respuesta simpaticomimética, como la reacción de pelea o huída. Por ejemplo, la frecuencia cardíaca aumentará y las pupilas se dilatarán, se mobilizará la energía corporal y la sangre fluirá a órganos esenciales.
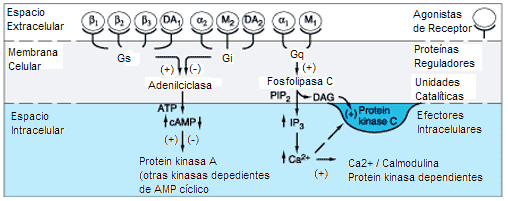
Existen varios tipos de receptores adrenérgicos divididos en dos grupos principales, los receptores alfa (a) y los receptores beta (ß).[2] * Receptores a: se unen con epinefrina y norepinefrina. La fenilefrina es un agonista selectivo del receptor a.[3] Existen dos subtipos, el receptor a1 y el receptor a2. * Receptores ß: asociados a proteínas G y que activan a la adenil ciclasa. Los agonistas que se unen a los receptores ß producen un incremento en la concentración intracelular del segundo mensajero AMPc.
La epinefrina actúa tanto con los receptores a y ß causando vasoconstricción y vasodilatación respectivamente.[4] Aunque se sabe que los receptores a son menos sensibles a la epinefrina, cuando se activan, se sobreponen a la vasodilatación mediada por los adrenoreceptores ß. El resultado es tal que a concentraciones circulantes elevadas de epinefrina causan vasoconstricción. A niveles circulantes bajos de epinefrina, la estimulación de receptores ß predomina, produciendo vasodilatación general. El mecanismo de la acción de los receptores adrenérgicos se fundamenta en la molécula acoplada al receptor en el lado intracelular de la membrana plasmática. Los ligandos para el receptor a1 y el receptor a2 son la adrenalina y la noradrenalina. Un receptor a1 tiende a unirse a una proteína Gq, resultando en un incremento del Ca2+ intracelular causando así la contracción de la musculatura lisa. Los receptores adrenérgicos a2, a su vez, se unen con una proteína Gi, que reduce la actividad del AMPc, produciendo así la contracción del músculo liso. Los receptores ß se unen a la proteína Gs[2] y aumenta la actividad intracelular del AMPc, resultando en contracción del músculo cardíaco, relajación del músculo liso y glucogenólisis.
***Los Fosfolípidos De Inositol y sus Segundos Mensajeros: Ip3 y dag
Los fosfolípidos de inositol (PI) son compuestos minoritarios de las membranas biológicas. Estas moléculas pueden ser fosforiladas bajo estímulo por kinasas específicas hasta fosfatidilinositol 4,5-bifosfato (PIP2) (Fig. 6). La activación de estas kinasas puede ser mediada tanto por GPCRs como por RTK, sin embargo nosotros nos centraremos en la mediada por los primeros. Cuando la hormona entra en contacto con el receptor (p.ej. receptores a-adrenérgicos), éste puede contactar con la proteína G inactiva y activarla. La Ga0 activa tiene capacidad para contactar y activar una proteína anclada a la cara interna de la membrana plasmática; la fosfolipasa C (PLC). Existen varias isoformas de PLC: la PLC-p , la PLC-y y la PLC-8 . La PLC-p es la que se activa en este caso que nos ocuupa. El sustrato de la PLC-p es el PIP2. El enzima hidroliza este fosfolípido justo en el enlace éster que conecta el fosfato con el glicerol, de esta acción resultan dos productos; el Diacilglicerol (DAG) y el Inositol 1,4,5 Trifosfato (IP3). El DAG permanece en la capa interna de la membrana, mientras el IP3 que es una molécula hidrófila difunde al citosol celular, donde va a actuar como ligando regulador de un canal de calcio (Ca+2), se une a este canal abriéndolo y el Ca+2 sale del retículo endoplasmático u otras vesículas intracelulares que almacenen este ión, hacía el citosol. La subida momentánea del Ca+2 citosólico va a promover la incrustación de un enzima, la PKC (Proteína Kinasa dependiente de Calcio), a la cara citosólica de la membrana plasmática (Figura 7). En esta situación, la PKC puede ser activada por el DAG y de esta manera fosforilar a sustratos proteicos específicos en residuos de serina o treonina.
Otra transducción de la señal: Ejemplo de receptores b-adrenérgicos unidos a proteínas G estimulando a la Adenilato Ciclasa (Gs). Verlo en transducción apartado antipsicóticos.
Fuentes:
ESTIMULANTES (Simpaticomiméticos):
Anfetamínicos:
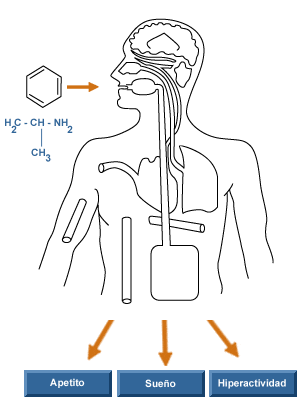
Las anfetaminas constituyen un grupo de substancias semejantes estructuralmente a la adrenalina y a la efedrina. La molécula representativa del grupo es la anfetamina. Es una amina simpaticomimética indirecta, que además de ejercer efectos sobre los receptores noradrenérgicos a y b periféricos tiene claros efectos sobre el sistema nervioso central: estimula el centro respiratorio bulbar, actua sobre la corteza cerebral y parece también estimular el sistema activador de la formación reticular. También disminuye el efecto depresivo de otras drogas sobre el sistema nervioso central.
No Anfetamínicos:
En este grupo también se engloban algunas sustancias con acción selectiva de inhibición de la recaptación de la noradrenalina y dopamina con efectos similares a las anfetaminas, ver la sección antidepresivos de esta web.
Efectos Secundarios
El aumento de la actividad psíquica se manifiesta por un aumento de la capacidad de concentrarse en tareas concretas. La realización de test a estudiantes nos indica como bajo el efecto de anfetaminas se aumenta la capacidad de respuesta, sin embargo aunque contestan mayor número de respuestas también cometen un mayor numero de errores. Euforia: se produce un aumento de la atención, mayor capacidad de comunicación, aumento de actividad. Disminución de la sensación de fatiga: la fatiga es un mecanismo regulador del organismo mediante el cual se frena la actividad. El hecho de que la sensación de fatiga este disminuida no significa que esta no se presente, con el consiguiente riesgo para la persona que no sabe dosificar su esfuerzo y puede terminar en un agotamiento agudo con serias consecuencias para su salud. Insomnio: la anfetamina retrasa la aparición de sueño, pero no de manera indefinida, por lo que al suspender la administración de las anfetaminas aparece como fenómeno de rebote un sueño mas profundo y una necesidad imperiosa de dormir.
El patrón del sueño se altera y puede tardar varios meses en volver a la normalidad. Disminución del apetito. Debido a esta acción en ocasiones se utilizan en el tratamiento de la obesidad, aunque esta acción es muy discutible. Aparece rápidamente tolerancia, siendo necesario aumentar la dosis para obtener el mismo efecto. Esto hace que con frecuencia las personas se hagan dependientes a las anfetaminas. Aparición de conductas estereotipadas, caracterizadas por la aparición en la persona que consume las anfetaminas de movimientos repetitivos. Al mismo tiempo pueden producirse sudoraciones, aumento de la frecuencia cardiaca, aumento de la presión arterial, sequedad de boca, vértigo, temblores. Generalmente estos efectos desaparecen al cabo de 3 ó 4 horas, apareciendo cansancio y en ocasiones la persona se siente deprimida e incapaz de concentrarse.
Fuentes:
- http://wapedia.mobi/en/Stimulant
- http://www.estafilococo.com.ar/estimulantesnc.htm
- http://www.camporenacimiento.com/adiccion/anfetaminas.htm
- http://www.arconet.es/familia/111Lavidahumana/Droga/Anfetaminas.htm
NO ESTIMULANTES (Parasimpaticomiméticos):
Agonistas del receptor de la Noradrenalina a2:
El uso principal de esta medicación es el tratamiento de la hipertensión. Actúa estimulando ciertos receptores cerebrales (de tipo alfa-adrenérgico) que a su vez relajan los vasos sanguíneos de otras partes del cuerpo, haciendo que se ensanchen. Tiene especificidad por los receptores presinápticos α2 en el Centro Vasomotor del SNC. Esta unión inhibe la producción de Norepinefrina, disminuyendo de ese modo la actividad simpática, predominando la actividad parasimpática.
Estimula selectivamente los receptores cerebrales que actúan como sensores de los niveles sanguíneos de catecolaminas. Estos receptores cierran un bucle de retroalimentación negativo que comienza con las órdenes de los nervios simpáticos que descienden del cerebro que controlan la producción de catecolaminas epinefrina también conocida como adrenalina, y norepinefrina en la médula adrenal. Confundiendo al cerebro de modo que este crea que los niveles de catecolaminas son mayores de lo que en realidad son, la clonidina hace que el cerebro reduzca sus señales hacia la médula adrenal, lo que a su vez baja la produción de catecolaminas y los niveles sanguíneos. El resultado es una tasa cardiaca y presión sanguínea menores, con el efecto secundario de la sequedad bucal y la fatiga. Si se deja de administrar repentinamente la clonidina el sistema nervioso simpático volverá a producir altos niveles de epinefrina y norepinefrina, incluso más altos que antes del tratamiento, provocando una hipertensión de rebote. Este rebote se puede evitar retirando paulatinamente el tratamiento.
BETA BLOQUEANTES:
Farmacodinamia:
Actúan con afinidad preferentemente sobre los receptores cardíacos b1, aunque también tienen afinidad para los receptores vasculares periféricos o bronquiales b2.
Reduce la actividad simpática, por lo que se indica en la Fobia Social. De esta forma el paciente percibe menos el ritmo cardíaco (palpitaciones) y tiende a disminuir el circuito de retroalimentación que se produce en este trastorno: miedo-->palpitaciones-->más miedo.
Los betabloqueadores son agentes antagonistas competitivos de acciones norepinefrínicas y epinefrínicas en los receptores beta adrenérgicos.
El propanolol es un bloqueador beta no selectivo; su efecto puede ser a través de receptores beta1 o beta2, centrales o periféricos. El propanolol se concentra en el cerebro debido a su alta liposolubilidad (Lipinski J. et al. 1984).
Usos en Psiquiatría
Disquinesia Tardía
Síndrome Extrapiramidal
Síndrome de las Piernas Inquietas
Taquicardia
Furia Explosiva
Conducta Violenta
Agresión
Ansiolítico
Algunos observadores han reportado acerca de la utilidad del propanolol en la disquinesia tardía y en el síndrome extrapiramidal (Bacher N., Lewis H. 1980; Kulik F., Wilbur R. 1980. citados por Chaudhry R. et al. 1982). Chaudhry R. et al (1982) describen el caso de una mujer con diskinesia y síndrome extrapiramidal, para quien el propanolol fue efectivo y mejoró ambos trastornos, a una dosis de 60 mg./día. El sistema específico betabloqueador puede influenciar al sistema reticular de activación, el cual es esencialmente adrenérgico. Esto puede resultar en una modificación de los niveles de estimulación, lo que puede influenciar la actividad motora anormal. Un segundo mecanismo postulado es sobre las neuronas de los ganglios basales, al normalizarse el funcionamiento de las membranas neuronales.
El síndrome de las piernas inquietas es un trastorno idiopático, semejante a la acatisia que se ha reportado, que responde al tratamiento con propanolol, también se ha encontrado gran beneficio al tratar la acatisia inducida por neurolépticos; también hay disminución muy importante del temblor por litio; parece ser que el propanolol ejerce su efecto beneficioso sobre la acatisia a través de bloqueo beta2 periférico o central (Lipinski J. et al. 1984).
La acatisia puede responder al propanolol, existen investigaciones que indican que puede llegar a ser considerado como tratamiento de elección (Janicak P. 1993).
En el tratamiento con litio, el temblor puede aparecer como una complicación, puede ser manejado con propanolol (30–120 mg), se indica dos horas antes de la necesidad de estabilizar las manos (Janicak P. 1993).
He tenido la experiencia en nuestros pacientes hospitalizados, de lograr control efectivo de la taquicardia como efecto secundario de los neurolépticos, al administrar pequeñas dosis de propanolol (10 a 30 mg/día).
Se ha reportado la efectividad del propanolol en el tratamiento de la furia explosiva y de la beligerancia episódica, en pacientes con daño cerebral agudo. En altas dosis (320 a 520 mg./día.) reduce la sobreagresividad, la violencia y la furia en pacientes esquizofrénicos, con síntomas floridos, que no habían remitido con los tranquilizantes mayores (Yudofsky S. and al. 1981).
En los pacientes con psicosis de Korsakof asociada con rabia severa, agitación, conducta violenta, que no responden a intervención psicofarmacológica y conductual tradicional, se inicia tratamiento con dosis altas de propanolol, hasta 600mg./día, en 4 dosis. Dosis por encima de 20 mg./día se han usado para tratar la agresión y otros síntomas relacionados con la esquizofrenia y los trastornos maníaco-depresivos. El propanolol no tiene propiedades antipsicóticas aunque sí tiene propiedades anticonvulsivas (Yudofsky S. et al. 1984).
Los betabloqueadores tienen efectos ansiolíticos; se ha pensado que esto se debe a los efectos periféricos más que a los centrales, con los siguientes argumentos: 1) Se piensa que los betabloqueadores no pasan la barrera hematoencefálica para ejercer su efecto ansiolítico. 2) Componentes con una acción central similar, pero sin rol betabloqueante periférico, no reducen la ansiedad. 3) Los efectos Centrales de estas drogas no pueden ser detectados a las dosis administradas normalmente para aliviar la ansiedad (Hartley R. et al. 1983). Se considera que el efecto ansiolítico del propanolol, se ejerce impidiendo que los receptores adrenérgicos periféricos respondan a las manifestaciones concomitantes del sistema nervioso autónomo, que provoca la ansiedad originada centralmente (Gottschalk L. 1982). Los pacientes que se encuentran en situaciones agudas de estrés (discursos, exámenes, entrevistas de trabajo), en los cuales no sean deseables las posibles alteraciones psicomotoras o intelectuales producidas por las benzodiazepinas, pueden ser candidatos para el bloqueo farmacológico beta (20 a 40 mg. poco tiempo antes del evento) (Greenblatt D. y Shader R. 1982).
Estudio comparativo de betabloqueantes y placebo respecto al tratamiento de la acatisia
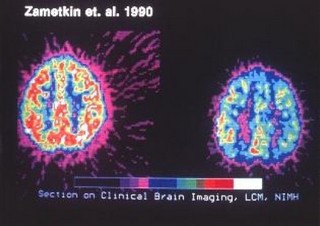
Cerebro normal a la izquierda cerebro TDAH a la derecha
El funcionamiento de los lobulos frontales se asocia con los niveles mas elevados de la funcion cortical, entre los cuales se encuentran los inherentes a la actividad intelectual. Zemetkin y colaboradores, demostraron un aprovechamiento menor de glucosa en areas cerebrales frontales a traves de los estudios con tomografia con emision de positrones.

Estos tomografía por emisión de positrones (PET) muestran que los pacientes con TDAH tenían niveles más bajos de los transportadores de dopamina en el núcleo accumbens, una parte del centro de recompensa del cerebro, que los sujetos control.
"Estos hallazgos sugieren que una variable adicional en relación con los transportadores de dopamina serían necesarios para dar cuenta de la gravedad de los síntomas de falta de atención en el TDAH", dijo Volkow.
"Especulamos que esta variable puede ser otros niveles más bajos de la liberación de dopamina en sujetos con TDAH".
Si los sujetos con TDAH liberan menos dopamina para empezar, puede terminar con los niveles más bajos de los transportadores de la dopamina como resultado de la regulación a la baja - es decir, el intento del organismo para compensar los bajos niveles de dopamina, reduciendo el número de proteínas de la recaptación.
Esto explicaría también por qué los sujetos de control, con mayores niveles de liberación de dopamina, tuvieron puntuaciones más bajas de falta de atención que los sujetos con TDAH con niveles similares de los transportadores de dopamina.
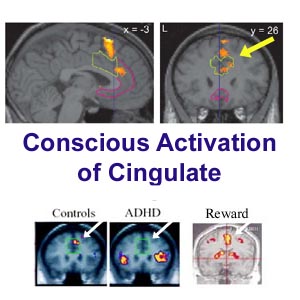
La región del estriado es una red de estructuras en el cerebro medio que motiva a las personas a adoptar un comportamiento agradable o gratificante.
El cíngulo anterior es mayor en el cerebro, y se activa normalmente cuando uno se obtiene la recompensa esperada.
Theodore Beauchaine, profesor asociado de psicología de la UW y autor principal del artículo, afirma que este proceso se produce no lo hace por lo menos tan rápidamente, en niños con déficit de atención hiperactividad o trastornos de la conducta.
"Cuando los niños se involucran en conductas impulsivas que buscan estimular a sí mismos y divertirse", dijo Beauchaine. "Los niños con trastorno de hiperactividad y déficit de atención están buscando siempre para divertirse y eso es lo que los mete en problemas", agregó.
Pero incluso cuando la recompensa para los niños con estos trastornos, se seguirán centrándose en la recompensa mucho tiempo después y la corteza cingulada anterior no parece que se active.
Para el estudio, los investigadores compararon 19 niños con uno o ambos trastornos y 11 niños de desarrollo normal. Los adolescentes con edades comprendidas entre 12 a 16.
Se les pidió que jugaran un juego simple que a veces les daba una recompensa monetaria por las respuestas correctas.
Beauchaine dijo que no hubo diferencias en la exactitud o la velocidad - la respuesta conductual - entre los dos grupos.
Pero había una diferencia en la activación cerebral.
"Esto muestra que hay una anormalidad, pero no en el lugar que esperábamos encontrar. Esperábamos encontrar una diferencia en el funcionamiento del cuerpo estriado, pero en cambio se encuentra en funcionamiento cingulada anterior".
Todo Neuroimagen del TDAH (SPECT, PET-TAC)
Etiología posible del TDAH
UNA APROXIMACIÓN NEUROBIOLÓGICA Se podría definir la atención como la capacidad de escoger entre un conjunto de estímulos, el que sea de verdadero interés. En los seres humanos las funciones de la atención se distribuyen en un sistema posterior que se orienta a estímulos novedosos y un sistema anterior que se encarga de las funciones ejecutivas. El sistema posterior, que incluye la corteza parietal, los colículos superiores y el núcleo pulvinar, recibe inervación noradrenérgica procedente del locus coeruleus. La noradranalina (NA) inhibe la descarga espontánea de las neuronas que incrementa la tasa de estímulo relevante/ruido de las células blanco y logra que el sistema posterior se oriente e involucre en un estímulo novedoso. En ese momento la función de atención pasa al sistema anterior ejecutivo, integrado por la corteza pre-frontal (CPF) y la circunvolución anterior del cíngulo. La reactividad de la CPF y el cíngulo anterior a las señales que ingresan está modulada primariamente por las aferencias dopaminérgicas procedentes del área ventral del tegmento mesencefálico. La fibras ascendentes estimulan los receptores D1 post-sinápticos en las neuronas piramidales en la CPF y el cíngulo anterior las cuales a su vez, facilitan la llegada a los receptores excitatorios N-metil-D-aspartato (NMDA) de las aferencias provenientes del sistema posterior de atención. De este modo, la dopamina (DA) permite la entrada selectiva a la CPF y al cíngulo de las aferencias excitatorias reduciendo la actividad neuronal irrelevante de las neuronas durante el desempeño de una función ejecutiva. En condiciones normales, los transportadores dopaminérgicos la absorben para que luego se pueda reutilizar.
http://colombiamedica.univalle.edu.co/Vol38No4/html%20v38n4/v38n4a14.html
***
Sabemos que estímulos externos activan tanto el sistema central de norepinefrina como el sistema periférico de tipo simpático. De igual forma, desde un punto de vista neuroanatómico sabemos que los centros de atención en el cerebro que se encuentran tanto en el área posterior del cerebro (encargada de cambiar de estímulos no significativos a significativos) como la anterior o frontal (la parte “ejecutiva” de la atención) utilizan las catecolaminas como un ingrediente esencial en la transmisión de mensajes. Se sabe que el sistema frontal está regulado por la dopamina (DA) desde el núcleo VTA (Ventral Tegmental Area), mientras que el sistema posterior está regulado por la norepinefrina (NE) desde el núcleo LC (Locus Ceruleus).(37) Conectando ambos núcleos hay fibras con alto contenido de serotonina (5HT).
Más interesantes, sin embargo, son los estudios de asimetría en el núcleo caudal por medio de imágenes de resonancia magnética (MRI). Mientras que un 72% de niños normales mostraron una asimetría donde el núcleo caudal derecho es más grande que el izquierdo, un 63% de los niños con hiperactividad mostraron una correlación inversa, esto es, el núcleo caudal izquierdo era más pequeño que el derecho.(28) Esto concuerda con la teoría asimétrica de control del cerebro, donde la dopamina es más activa en el lado izquierdo mientras la norepinefrina lo es en el lado derecho. De los dos neurotransmisores, se cree que la dopamina juega un papel más importante en el trastorno.
Estructuras anteriores del corpus callosum son más pequeñas en los niños con el diagnóstico cuando se comparan con niños sin la condición.
***
Si bien se sabe que las neuronas dopaminérgicas de la sustancia negra son los principales moduladores de la función de los ganglios basales mediante la liberación de dopamina en la sinapsis distal con las neuronas del estriatum, globo pálido y subtálamo, también se sabe que la liberación local de dopamina hiperpolariza la neurona presináptica de la sustancia negra mediante la liberación dendrítica de dopamina, lo que conduce primariamente a la auto-inhibición de la neurona dopaminérgica (Falkenburger y col. 2001). Los especialistas concuerdan en que el polimorfismo confiere al receptor una especie de sub-sensibilidad a la dopamina endógena. Al disminuir el efecto inhibitorio el resultado final es un estado de excitación motora por arriba de lo normal. En este sentido, el incremento en los niveles de dopamina que produce la administración de metilfenidato sería el mecanismo mediante el cual se compensaría la deficiente actividad del DRD4 lo que se observa por el efecto calmante de esta anfetamina en el paciente con TDAH (Barr, 2001). Evidencia de este mecanismo de subsensibilidad a la dopamina es proporcionada por el estudio de Ernst y col. (1999). Mediante la administración de un análogo de la dopa, el trazador [(18)F]DOPA a un grupo de 10 niños con TDAH y 10 controles, y su seguimiento con la técnica de tomografía de emisión de positrones, se demostró que el grupo con TDAH presentaba una acumulación significativamente mayor (48%) del trazador en la región medial-cerebral, que incluye los cuerpos celulares de las neuronas ricas en dopamina de la sustancia negra y del tegmentum ventral. El resultado sugiere una mayor actividad de la enzima dopa-descarboxilasa y consecuentemente una mayor síntesis de dopamina en esa región, lo cual, fisiológicamente, ocurre en respuesta a niveles extracelulares bajos de dopamina y al bloqueo de los receptores de la misma. Esto concuerda con la teoría de subsensibilidad del receptor; esta subsensibilidad conduce a mecanismos de transmisión y señalización inadecuados en la neurona postsináptica, que mediante un sistema de retroalimentación negativa estimula al cerebro medial a producir más dopamina. (Ernst y col. 1999).
***
La respuesta tiene dos vertientes. En primer lugar, la administración de estimulantes, o cocaína, ejerce un efecto calmante, similar al visto en humanos con TDAH y, al igual que en humanos normales, la droga provoca hiperactividad al ser administrada en ratones normales. En segundo lugar, los niveles cerebrales extracelulares de dopamina se incrementan según lo esperado cuando la droga se aplica a ratones normales. Sin embargo, la administración en el ratón knockout, no produce ningún cambio en tal medio, lo que sugiere firmemente que los estimulantes no afectan al sistema de la dopamina en este ratón, y por lo tanto ejercen su efecto calmante posiblemente a través de la modulación de otros neurotransmisores afectados por estas drogas. Para probar esta hipótesis se evaluó un inhibidor selectivo del transportador de norepinefrina (NE): nisoxetina: no hubo ningún efecto sobre la hiperactividad, lo que, al menos en este ratón, sugiere que probablemente la NE no esté implicada en el mecanismo mediante el cual ejercen su efecto los estimulantes. Por el contrario, un inhibidor de la recaptura de serotonina, fluoxetina causó dramática reducción de la hiperactividad, al igual que lo hicieron otras drogas que incrementan la actividad de los receptores de serotonina (quipazine) o que incrementan los niveles cerebrales de serotonina (el precursor de serotonina triptofano y el 5-hidroxitriptofano). Las conclusiones a las que llega el autor son 1) la hiperactividad inducida por altos niveles de dopamina puede reducirse elevando el tono serotoninérgico 2) los estimulantes probablemente modulen la conducta aumentando los niveles de serotonina mas que inhibiendo la recaptura de dopamina (Gainetdinov y col. 2001).
***
La memoria de trabajo es la capacidad para mantener una representación mental para guiar la conducta. Es “recordar con el propósito de”. Frecuentemente el recordar, debe esperar un periodo de tiempo hasta que los eventos externos asociados ya no estén presentes. Inhibición de respuesta es la capacidad para retrasar una respuesta a un evento ambiental inmediato. La AR es prácticamente imposible sin este retraso que permite el reforzamiento de la respuesta. Además permite la interiorización de las demás FE, suprimiendo los movimientos músculo-esqueléticos asociados con la dirección de la conducta de las FE (Barkley, 2000). La mayor información acerca de la localización de estas funciones proviene del estudio con ratas y primates (Arrnsten y col. 1996). La depleción de NE en primates, inducida experimentalmente o debido a la edad, produce déficit de la memoria de trabajo, incrementando la distractibilidad. En estos animales este déficit puede mejorarse con la administración de agonistas del receptor a2 de NE como la clonidina y la guanfesina, y se agrava con los antagonistas a2 pero no con los a1. Tres subtipos de receptores a2 en humanos se han descubierto y clonado a la fecha: a2A, a2B, y a2C. El efecto final de administrar un agonista, corresponde al sitio(s) de mayor concentración del subtipo de receptor dentro del encéfalo. Así, se ha encontrado que el subtipo a2A presenta la mayor densidad de influencia en la CPF, incluyendo el área 46, y en el locus coeruleus. De esta forma, en primates viejos, la guanfesina demostró que mejora la ejecución de una prueba de respuesta retrasada, sin los efectos sedativos e hipotensores que acompañan a la clonidina. Resultados similares se replicaron en primates jóvenes. Los agonistas a2 se muestran más efectivos bajo condiciones de alta interferencia o distracción, cuando la CPF esta bajo presión. Además, aún dosis muy bajas de guanfesina pueden proteger la actividad de los primates del efecto negativo de estímulos irrelevantes. De esta forma, la teoría de la depleción de NE ha encontrado fuerza en experimentos clínicos en humanos, al encontrar que los agonistas de NE mejoran las funciones cognoscitivas de la CPF. La clonidina ya ha sido probada en el TDAH, pero sus efectos hipotensivos y sedativos han limitado considerablemente su utilidad clínica. La guanfesina se ha iniciado en ensayos abiertos con pacientes con TDAH con resultados alentadores (Arnsten y col. 1996). Sin embargo la participación de la CPF en el deterioro de las funciones cognoscitivas y afectivas del TDAH no puede atribuirse únicamente a una deficiente actividad de los receptores a2. Existe evidencia de que la estimulación de los receptores a1 interfiere con la función de la CPF a través de la activación de la vía del segundo mensajero intracelular fosfatidil-inositol-proteincinasa-C. La sobreactividad de este segundo mensajero también ha sido ligada a la manía, un trastorno que comparte algunas similitudes con síntomas del TDAH. Por lo tanto se trata mas bien de una transmisión alterada del sistema de NE la que contribuye a los síntomas del TDAH: deficiente estimulación a2 y/o excesiva estimulación a1 (Arnsten, 2000).
Fuentes:
- http://www.bnl.gov/bnlweb/pubaf/pr/PR_display.asp?prID=998
- http://www.bnl.gov/bnlweb/pubaf/pr/PR_display.asp?prID=06-124
- http://www.topnews.in/brain-abnormality-linked-adhd-identified-2141361
- http://www.psyncron.com/es/docs/tratatmientotdah.pdf
- http://www.tendencias21.net/Los-ninos-con-deficit-de-atencion-tienen-un-cerebro-distinto_a4909.html
- http://rehab-almenara.org/academic_archivos/presenta_2008/Hiperactividad.pps
- http://psicofarmacologia.info/Tninez.html
- http://bloguit.com/psicopedaloquiando/2008/01/12/
- http://www.pediatriaatlantico.org/Asambleas/hiperactividad.ppt
- http://www.alcmeon.com.ar/5/18/a18_05.htm
- http://www.tdah-andalucia.es/TDAH/funcionesejecutivas1.pdf
- http://cyberpediatria.com/sddacanizales.pdf
- http://www.aepij.com/aepnya/revista/2002_01/2002(1)82-88.pdf
- http://sanliz.com/nooanalepticos
- http://sanliz.com/anfetaminas
- http://espanol.geocities.com/aguilera99/volumen49.htm (el mejor)
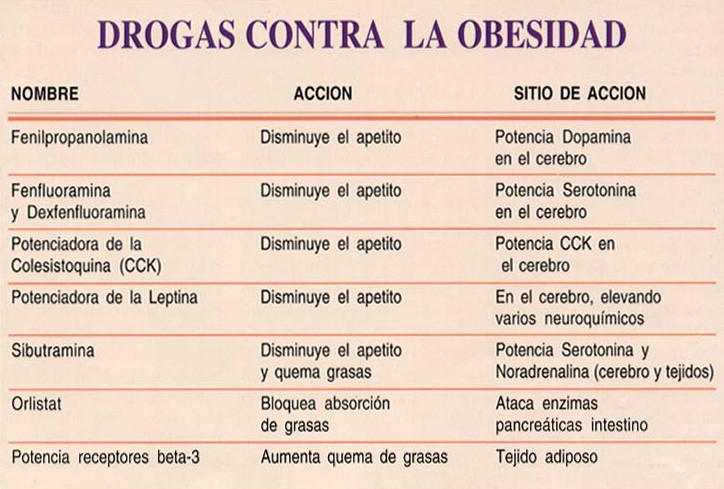
ANOREXÍGENOS: (Retirados, en algunos países todavía se comercializan.)
Disminuyen el apetito
Noradrenérgicos:
Los anorexígenos noradrenérgicos actúan a nivel del centro del apetito y presentan acción estimulante del SNC y riesgo de producir adicción. Por ello, han sido recientemente retirados del mercado farmacéutico en España y otros países de la Unión Europea. En cualquier caso, al cabo de 3-8 semanas pierden su acción supresora del apetito (por depleción total de las catecolaminas neurotransmisoras en el hipotálamo).
Serotoninérgicos:
Los anorexígenos serotoninérgicos actúan a nivel del centro de la saciedad, carecen de acción estimulante y no son susceptibles de abuso. Su acción se puede mantener durante un año. Pueden producir hipertensión pulmonar primaria. Además se han detectado ciertas anomalías cardíacas (valvulopatías) en pacientes con tratamiento conjunto de fenfluramina y fentermina.
Hasta ahora los fármacos más utilizados han sido los anorexígenos (medicamentos que reducen el apetito), que se clasifican en: noradrenérgicos y serotoninérgicos.
Varios fármacos se clasifican específicamente como anorexígenos, o sustancias que suprimen el apetito. Se recetan para el tratamiento de la obesidad y, como las anfetaminas, estimulan el Sistema Nervioso Central. Solamente deben recetarse durante períodos cortos, ya que con un uso continuado se desarrolla tolerancia. Su consumo se ha detectado, de forma incontrolada, entre adolescentes que quieren adelgazar y caen en la anorexia.
Medicamentos anorexígenos |
|||
| Nombre genérico | |||
| Agentes noradrenérgicos | Benfetamina | ||
| Fendimetrazima | |||
| Dietilpropion | |||
| Fentermina | |||
| Mazindol | |||
| Fenilpropanolamina | |||
| Agentes serotoninérgicos | Fenfluramina | ||
| Dexfenfluramina | |||
| Fluoxetina | |||
| Efedrina/cafeína | |||
* DEA: Drug Enforcement Agency (USA). |
|||
Potenciales contraindicaciones del uso de medicamentos anorexígenos |
| - IMC < 27,8 |
| - Edad < 18 años > 60 años |
| - Embarazo o lactancia (contraindicados absolutamente) |
| - Uso frecuente de otros agentes anorexígenos |
| - Hipertensión no controlada (sistólica >165 ó diastólica >100 mmHg) |
| - Enfermedad cardiaca no controlada |
| - Glaucoma no controlado |
| - Insuficiencia renal o hepática |
| - Historia de abuso de drogas |
| - Uso frecuente de fármacos antidepresivos (litio, fluoxetina, inhibidores de MAO) |
| - Historia de enfermedad maníaco depresiva o depresión mayor |
| - Hipertensión pulmonar |
| - Anestesia general en las últimas 2 semanas |
Fuentes
- http://doctorcesareo.blogspot.com/ (Si estás gordo lee esto, buenos consejos)
- http://www.scielo.org.ve/scielo.php?pid=S0798-02642007000100003&script=sci_arttext
- http://escuela.med.puc.cl/publ/boletin/obesidad/TratamientoFarmacologico.html
- http://www.tuotromedico.com/temas/farmacos_antiobesidad.htm
- http://psicofarmacologia.info/Tingesta.html
- http://www.arconet.es/familia/111Lavidahumana/Droga/Anfetaminas.htm
- http://www.obesos.org/
- http://www.gordos.com/
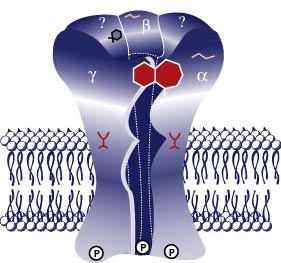
El receptor GABAA es una glicoproteína transmembrana compuesta de 4 subunidades, alfa, beta, gamma y delta, reconocidas en la actualidad. Es sensible a muscimol (agonista) como a la bicuculina y la picrotoxina (antagonistas). Existen varios tipos de receptores GABAA, diferentes entre ellos por algunos de sus subunidades. En este momento hay 6 subtipos de subunidades alfa, 3 subtipos de subunidades beta, 3 subtipos de subunidades gamma y 1 subtipo subunidades delta. Esto conduce no sólo una gran diversidad de estructuras sino también una acción farmacológica heterogenea, cuyas consecuencias son aún poco conocidos.
ANTIGABA:Fármacos convulsivos o analépticos. Ejercen su acción estimulante por medio del bloqueo de los mecanismos inhibidores, principalmente del Ácido Gamma-Aminobutirico (GABA). Los principales representantes de este grupo son: Picrotoxina , es un alcaloide que se encuentra en:
, es un alcaloide que se encuentra en:
La coca de Levante (Cocculus cocculus) es un arbusto originario de las regiones tropicales de Asia. El fruto de la coca de Levante es un fruto rojo que contiene la picrotoxina. La picrotoxina es un veneno neurotóxico que causa mareos, vómitos, espasmos musculares e incluso la muerte.
Bicuculina y Pentílenotetrazol.
ANTIBENZODIAZEPINAS:
El flumacenil, tiene afinidad por el receptor benzodiacepínico, pero carece de actividad, comportándose como un antagonista. Es soluble en agua; a diferencia del resto de las BZD es escasamente lipofílico y se une en bajo porcentaje a las proteínas plasmáticas ( 54-64% 7). Su comienzo de acción es rápido, teniendo una vida media de eliminación de 1 hora. Se metaboliza en el hígado y presenta el mayor aclaramiento de todas la BZD. Ejerce su efecto al antagonizar la acción de otras BZD, por lo que su administración de forma aislada (sin la acción de otras BZD) no muestra efectos fisiológicos
ANTIGLICINA:
Estricnina
La estricnina es un alcaloide de la nuez vómica y de otras especies del género Strychnos. La estricnina es un antagonista del aminoácido glicina, neurotransmisor de las células de renshaw. Al evitar la inhibición sobre las motoneuronas por parte de las células de renshaw, la estricnina produce hipercontracción muscular. Al inmovilizar el músculo del diafragma el individuo muere por asfixia.
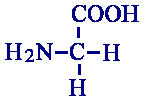
Neurotransmisor Glicina
La glicina tiene un receptor (distinto de los receptores para el GABA) que además puede unir ß-alanina, taurina, L-alanina, L-serina y prolina. No se activa con GABA. Un antagonista es el alcaloide estricnina, que bloquea la glicina y no interactúa con el sistema del GABA. Aumenta la conductancia para el cloro (parecido al receptor de la glicina al GABA-A). Este receptor se ha purificado utilizando el alcaloide estricnina. Este receptor es un complejo de subunidades a y ß, con estructura pentamérica, con homología al GABA-A y el receptor nicotínico. También posee 4 dominios transmembranales. En el citosol, se unen a gefinina para anclarse al citoesqueleto. Se piensa que otros receptores ionotrópicos pueden tener un sistema similar de anclaje a la membrana.

Como remedio múltiple, la nuez vómica se ha utilizado principalmente para el tratamiento de trastornos relacionados con el abuso de drogas narcóticas, alcohol, café o tabaco, consumo excesivo de alimentos grasos y bebidas y agotamiento mental en razón al trabajo excesivo.

El ataque epiléptico es debido a la repentina aparición y difusión de una intensa estimulación eléctrica del cerebro. Las corrientes cerebrales patológicas pueden extralimitadas localmente (ataques focales) o aparecer sobre toda la corteza cerebral (ataques generalizados) diferenciándose el gran mal con espasmos generalizados y el pequeño mal cuya sintomatología depende de la edad.
El encéfalo epiléptico
Es importante destacar que la epilepsia no es una enfermedad sino un conjunto de síntomas que se manifiestan por crisis repetidas generadas por en las neuronas. Es un síntoma que puede ser resultado de muchas enfermedades diferentes en el encéfalo.
La causa del 30% de los casos se debe al grupo de epilepsia sintomática. Las causas pueden ser traumatismo del cráneo, una malformación del encéfalo, falta de oxigeno durante el nacimiento, un tumor encefálico, alcoholismo, meningitis etc. La crisis a menudo comienzan en un grupo pequeño de neuronas que rodean el área afectada antes de extenderse a áreas más grandes del encéfalo.
El 70% restante de las personas con epilepsia tienen la forma idiopatica o desconocida. Se presume que la causa son cambios químicos más difusos en el encéfalo. Las crisis se originan en el área central del encéfalo en interacción con la corteza cerebral. Las crisis afectan de inmediato a todo el encéfalo.
Muchas crisis dañan el encéfalo por sí solas, debido a los efectos posteriores de la crisis. Puede haber muerte celular por falta de oxigenación, por una concentración excesiva de glutamato, que puede producir un desequilibrio en el balance de sal del encéfalo, y especialmente el calcio extra presente puede dañar las células muy activas. Tambien puede producirse pequeñas hemorragias encefálicas como consecuencia de la presión elevada en los vasos sanguíneos que drenan la sangre del encéfalo etc.
El resultado de este daño puede ser un encéfalo que funciona mal con todos los síntomas de una reducción de las facultades intelectuales, es decir, demencia.
Las crisis epilépticas, cualquiera que sea su tipo, siempre son causada por una transmisión excesivamente enérgica de impulsos en el encéfalo. Lo característico de estos impulsos anormales es que no ocurren en el patrón habitual, se produce sincronización. Pueden abarcar un grupo mayor o menor de neuronas o incluso todas las neuronas de todo el encéfalo. Todavía se desconoce la razón precisa de por que ocurren estos impulsos sincronizados. En teoría, existen dos factores que podrían causarlos:
El primer factor es referido al neurotransmisor GABA: Si la red neuronal inhibitoria no funciona como debiera, las otras neuronas tienen vía libre y comienzan a trasmitir impulsos epilépticos descontrolados. Esto ocurre cuando la concentración del neurotransmisor inhibitorio GABA no es normal
La otra posibilidad es cuando los estimuladores son demasiado potentes. Esto podría suceder cuando el neurotransmisor excitatorio Glutamato es muy alta en el encéfalo.
Las crisis epilépticas
1.Crisis generalizadas
Las descargas anómalas que generan crisis generalizadas se originan en la porción central del encéfalo y se extienden a toda la superficie, en una interacción entre la corteza cerebral y el centro del encéfalo. Son espontáneas y siempre conducen a perdida de la conciencia
1.a. Convulsiones generalizadas: La persona cae al suelo sin advertencia. La respiración se detiene y los brazos y las piernas se tornan rígidos (fase tónica), luego de lo cual la persona comienza a temblar y sacudirse (fase clónica). Después la persona yace quieta, a menudo con el rostro de color azul, hasta que la respiración se normaliza. La mayoría de las personas caen en un sueño profundo después de una crisis. Algunas personas pueden perder el control vesical y rectal durante una crisis. Al despertar no tienen memoria de lo sucedido; a menudo se encuentran cansadas y pueden presentar cefalea y dolor muscular por todo el esfuerzo por el que han atravesado.
1.b. Ausencias: Son crisis menores, ocurren en varios tipos de epilepsia infantil, pero en raros casos tambien en adultos. La crisis se inicia sin advertencia y consiste en intervalos breves de perdida de conciencia durante los cuales el niño esta temporalmente desconectado. En contraste con todos los demás tipos de crisis, las ausencias ocurren con mucha frecuencia, a menudo varios cientos de veces por día. Las ausencias a veces pueden complicarse con convulsiones.
2. Crisis parciales
Estas son causadas por descargas eléctricas anormales en un área localizada del encéfalo. Hay dos tipos:
2.a. Crisis parciales simples
Son crisis causadas por descargas anormales localizadas que no afectan a la conciencia, aunque pueden evolucionar a crisis parciales complejas si se altera la conciencia. Las crisis parciales simples son iguales al aura; es decir, advertencias de que ha comenzado una crisis. El aura más habitual es una sensación poco característica en el cuerpo, que se origina en el estomago y llega a la cabeza, pero tambien puede comprender luz, sonido, olfato, gusto u otras sensaciones.
En algunas circunstancias las anomalías se originan en el área de la corteza cerebral que controla el movimiento de los músculos o el sentido del tacto o el dolor. Aunque las descargas eléctricas tambien pueden producirse en el lóbulo temporal y pueden producir síntomas muy diferentes.
Durante las crisis parciales simples pueden ocurrir fenómenos sensoriales que se recuerdan luego y que a menudo conducen a especular si el paciente esta loco. Se producen cambios por ejemplo: en la percepción del tiempo, en la percepción de la luz, el sonido y el espacio, en la percepción de escala, cuadros de sueño similar a la vida, una sensación de vacío interior, ansiedad pronunciada etc...
Nuevas investigaciones indican que las crisis son hereditarias en algunos casos.
2.b Crisis parciales complejas
Son crisis causadas por una descarga anormal localizada que conduce a una alteración de la conciencia. En este tipo de crisis las descargas anormales estan localizadas principalmente en los lóbulos temporales. Durante las crisis aparece automatismo, es común observar masticación, gesticulación y otros movimientos repetitivos y sin propósito, denominados estereotipias.
Tanto en las crisis parciales complejas como en las simples las descargas eléctricas pueden extenderse a todo el encéfalo. Esto se denomina generalización secundaria y conduce a que las crisis terminen con convulsiones generalizadas y perdida del conocimiento.
Estado de mal epiléptico
Es un estado en el cual una crisis sigue a la otra sin que la persona recupere la conciencia entre las crisis. Puede producirse como convulsiones repetidas, parciales o generalizadas, o solo como un estado constante de lejanía, en el caso de las crisis parciales complejas o las ausencias repetidas. El estado de mal epiléptico con crisis generalizadas es cuestión de vida o muerte.
Se necesita hospitalización inmediata y tratamiento para evitar las complicaciones, que consisten en la perdida de gran numero de células nerviosas y falta de oxigeno en el encéfalo.
Las causas más comunes consisten en mal tratamiento de una epilepsia ya conocida, una transición demasiado rápida de un fármaco a otro o, en algunos casos, las personas que olvidan ingerir su medicamento.
Epilepsia y Herencia
El riesgo de que los hijos de una persona epiléptica sufran epilepsia es de un 4% si se trata de epilepsia del padre es idiopatica. Si ambos padres padecen este tipo de epilepsia, el riesgo se eleva hasta un 20-30%.
Si un gemelo idéntico tiene epilepsia idiopatica, el riesgo de que el otro gemelo desarrolle epilepsia es del 80%. En gemelos no idénticos el riesgo es del 10-20%.
En la población general el riesgo de desarrollar epilepsia es de un 1%. En los miembros de la familia de las personas con epilepsia sintomática el riesgo es de un 2% mas o menos. Si ambos padres tienen epilepsia sintomática, existe cierta razón para tenerlo en cuenta. En estos casos, hay que considerar los tipos de crisis involucradas, ya que existe gran similitud en las familias respecto del tipo de crisis, el inicio de la epilepsia y los cambios en el EEG. Si ambos padres tienen un tipo leve, los problemas son menos graves.
http://html.rincondelvago.com/epilepsia_5.html
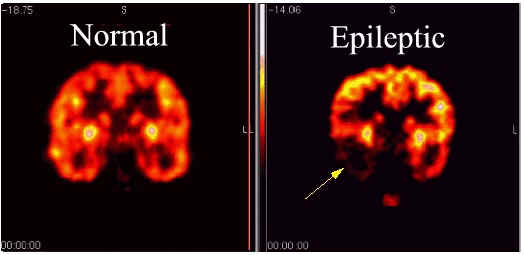
Epilepsia - El flujo sanguíneo y la captación de glucosa se miden por la PET con el fin de identificar los sitios ataques en el cerebro.En el 80% de los pacientes hay un aumento en el flujo sanguíneo y el metabolismo de la glucosa durante un ataque en la corteza cerebral. Sin embargo, entre crisis tiende a haber una menor captación de glucosa normal y el flujo sanguíneo.
Bomba de Calcio
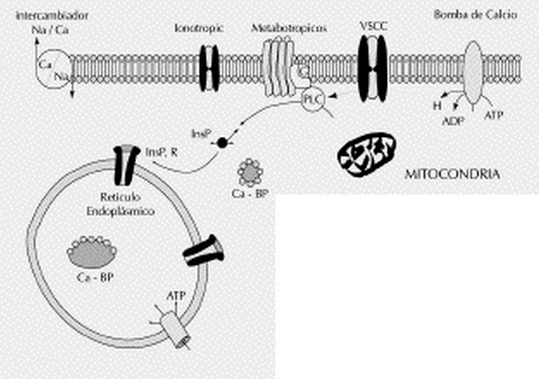
Es una proteína de la membrana celular de todas las células eucariotas. Su función consiste en transportar calcio iónico (Ca2+) hacia el exterior de la célula, gracias a la energía proporcionada por la hidrólisis de ATP, con la finalidad de mantener la baja concentración de Ca2+ en el citoplasma que es unas diez mil veces menor que en el medio externo, necesaria para el normal funcionamiento celula. En condiciones normales la concentración de calcio en el citosol es baja esto se logra mediante dos bombas de calcio, una en la membrana celular, que expulsa calcio hacia el exterior de la célula, ( permite que tres iones de sodio entren en la célula y con la energía que se libera sale un ion de calcio.) la otra introduce iones calcio a uno o mas organelos vesiculares internos de la célula. La proteína acarreadora atraviesa la membrana de lado a lado y actúa como ATPasa con capacidad para desdoblar ATP igual que ATPasa de sodio. Esta proteína tiene un sitio de unión para calcio en lugar de potasio.
Fuentes:
- http://www.monografias.com/trabajos5/memplas/memplas.shtml
- http://www.geosalud.com/NutricionOrtomolecular/nutricioncelular.htm
- http://sisbib.unmsm.edu.pe/BVRevistas/ciencia/v01_n1/calcio.htm
- http://escuela.med.puc.cl/publ/Cuadernos/2001/18.html
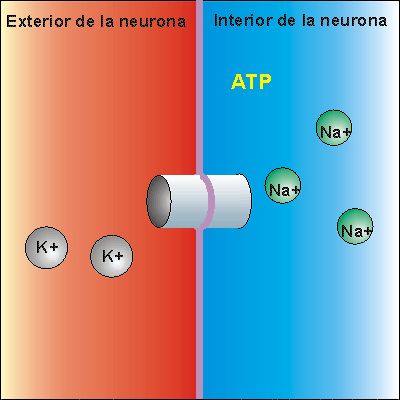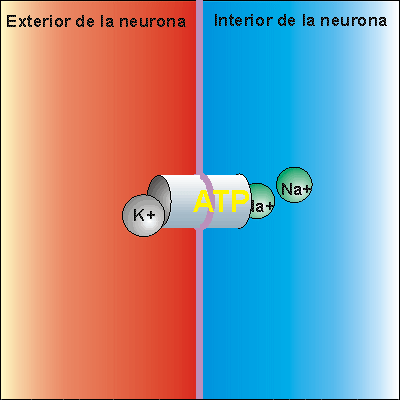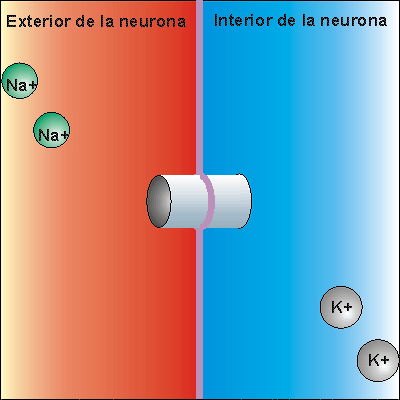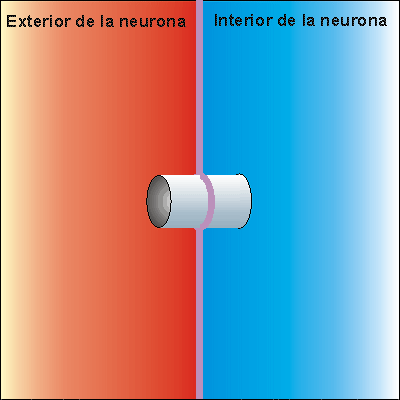
Bomba Sodio-Potasio
La bomba de Na+/K+ gasta ATP. Expulsa tres iones de sodio que se encontraban en el interior de la neurona e introduce dos iones de potasio que se encontraban en el exterior. Los iones sodio no pueden volver a entrar en la neurona, debido a que la membrana es impermeable al sodio. Por ello, la concentración de iones sodio en el exterior es elevada. Además, se pierden 3 cargas positivas cada vez que funciona la bomba de Na+/K+, aunque entren dos cargas de potasio. Esto hace que en el exterior haya más cargas positivas que en el interior, creando una diferencia de potencial. Se dice que la neurona se encuentra en potencial de reposo, dispuesta a recibir un impulso nervioso.
Cuando el impulso nervioso llega a una neurona en estado de reposo la membrana se despolariza, abriéndose los canales para el sodio. Como la concentración de sodio es muy elevada en el exterior, cuando los canales para el sodio se abren se invierte la polaridad, con lo que el interior de la neurona alcanza un valor electropositivo, respecto del exterior.
Si la despolarización provoca un cambio de potencial de 120 milivoltios más de los que tenía el interior se dice que se ha alcanzado el potencial de acción, que supone la transmisión del impulso nervioso a la siguiente neurona, ya que se crean las condiciones necesarias en el interior celular como para poder secretar neurotransmisor a la zona de contacto entre neuronas.
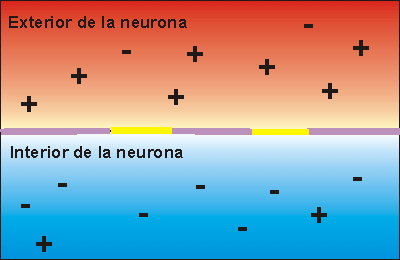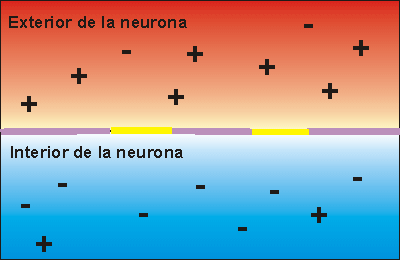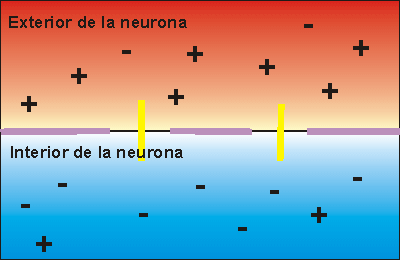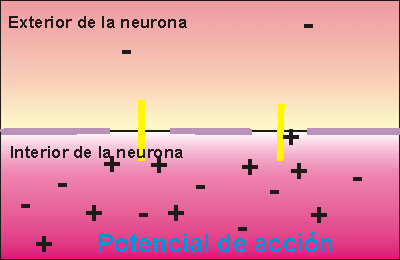
La transmisión del impulso nervioso sigue la Ley del todo o nada. Esto quiere decir que si la despolarización de la membrana no alcanza un potencial mínimo, denominado potencial umbral, no se transmite el impulso nervioso, pero, aunque este potencial sea rebasado en mucho, sólo se envía un impulso nervioso, siempre de la misma intensidad.
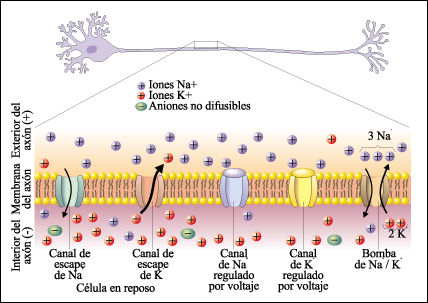
Canales ionicos de escape: Permaneces siempre abiertos y permiten que los iones se difundan a favor de su gradiante de concentracion Canales ionicos dependientedel voltaje: Se abren y se cierran en respuesta a los cambios de voltaje a traves de la membrana plasmatica Bomba de Sodio-Portasio: Mueve iones Na+ y K+ en contra de su gradiante de concentracion. transporta iones Na+ desde el interior y los intercambia por iones K+ del exterior.
Cuando la membrana celular se encuentra en potencial de reposo o polarizada, los canales de sodio regulados por voltaje presentan cerradas las compuertas que conectan con el medio extracelular y abiertas las compuertas intracelulares. Los canales de potasio regulados por voltaje, se encuentran cerrados. La despolarización se inicia cuando un estímulo desencadena el cambio de potencial de membrana: éste se torna positivo como consecuencia de la apertura de los canales de sodio. Alcanzado el umbral, muchos canales de sodio se encontrarán abiertos. El sodio difunde hacia el medio intracelular provocando la despolarización de la membrana y la lenta apertura de los canales de potasio. El potasio difunde hacia el medio extracelular. Por lo tanto, la despolarización es consecuencia de la difusión de sodio hacia el interior y de potasio hacia el medio extracelular. Cuando el potencial de membrana alcanza un máximo de despolarización, comienzan a cerrarse los canales de sodio disminuyendo la difusión de este ión. Los iones potasio continuan difundiendo hacia el medio extracelular pues sus canales permanecen abiertos. La salida de iones potasio, torna al potencial de membrana más negativo que el valor registrado en el estado de reposo. Recién después del cierre de los canales de potasio, el transporte activo de sodio y potasio restablece el potencial de reposo de la membrana.
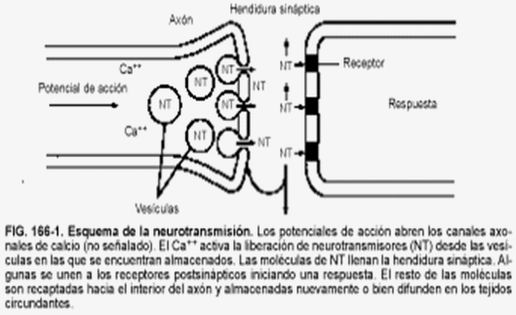
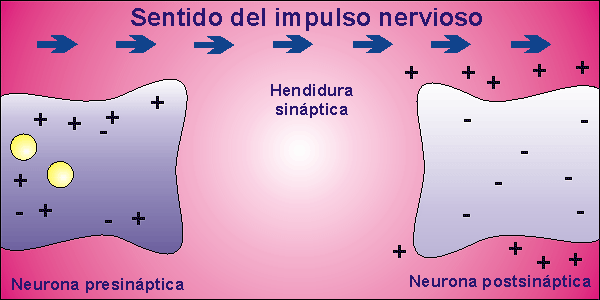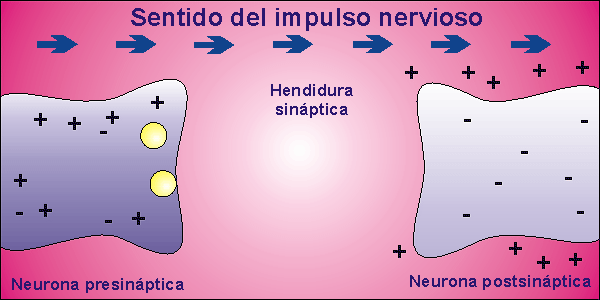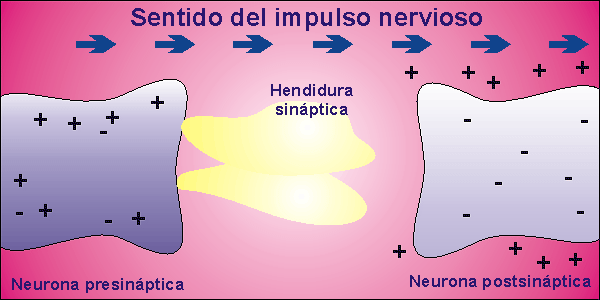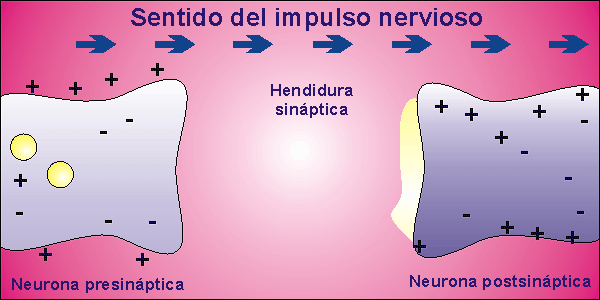
Sinapsis
Las neuronas, en la mayor parte de los animales, no se encuentran físicamente unidas. Existe un pequeño espacio entre ellas, llamado hendidura sináptica, al que se vierte el neurotransmisor desde la membrana presináptica, membrana de la neurona que envía el impulso nervioso, a la membrana postsináptica, membrana de la neurona que recibe el impulso nervioso. El neurotransmisor es la molécula responsable de despolarizar la membrana de la neurona que recibe el impulso nervioso, abriendo los canales para el sodio que permanecían cerrados.
Una vez que la neurona emite el impulso nervioso debe volver al inicial potencial de reposo. Para ello, la membrana se repolariza, cerrándose los canales para el sodio que estaban abiertos por la presencia del neurotransmisor. El neurotransmisor es destruido por acción enzimática y el potencial de reposo se alcanza al expulsar el sodio la bomba de Na+/K+.
- http://recursos.cnice.mec.es/
- http://www.monografias.com/trabajos41/potencial-membrana/potencial-membrana2.shtml
- http://es.wikipedia.org/wiki/Bomba_sodio-potasio
- http://html.rincondelvago.com/bomba-de-sodio-y-potasio.html
- http://www.unizar.es/departamentos/bioquimica_biologia/docencia/ELFISICABIOL/Canales/CanIonFB.htm
- http://www.oocities.com/siliconvalley/park/9312/canales.html
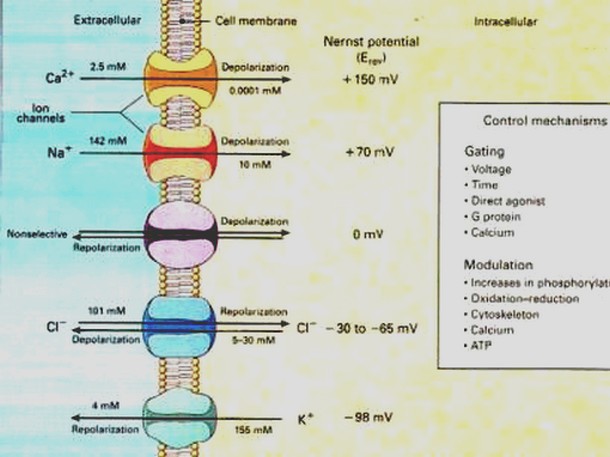
*mM (Micromoles), mV (Milivoltios). 1 mol es igual a 1000.000.000 nanomoles o 1.000.000 micromoles. 1000 mV es igual a 1 voltio.
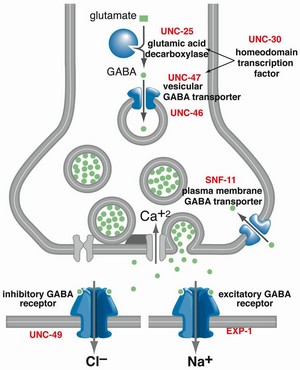
El receptor de GABA que pone excitatorio EXP-1 es un receptor que se encuentra en nematodos (gusanos) no en humanos. En rojo el gen que codifica la proteína.
Antiepilépticos/Anticonvulsivos: Gabapentina
Clasificación por farmacodinamia:
Bloqueantes de canales de Sodio:
Bloqueo de los canales de sodio dependientes de voltaje. Inhibe propagación del foco epiléptico. Son efectivos en ataques parciales y secundariamente en los generalizados, y en cuadros generalizados tónico-clónicos pero no en las ausencias.
Bloqueantes de canales de Calcio: Gabapentina
Bloqueo de los canales iónicos de calcio tipo N y L dependientes de voltaje. Se piensa que se une a la subunidad α2δ de los mismos en el sistema nervioso central. En la Etosuximida son de tipo T.
Agonistas del Receptor GABA:
Benzodiacepinas o Barbitúricos usados para la epilepsia, potencian el efecto del GABA y la apertura del canal al cloro. (Ver ansiolíticos).
Inhibidores de la recaptación del GABA:
Potencian el efecto del GABA al inhibir su recaptación por el GAT-1.
Inhibición irreversible de la GABA-Transaminasa:
La GABA Transaminasa (GABA-T) degrada el GABA a ácido succínico semialdehído que la Succínico semialdehído deshidrogenasa (SSADH) lo convierte a ácido succínico, inhibiendo esta enzima incrementamos la concentración del mismo en las sinapsis GABAérgicas
Bloqueantes de los receptores del Glutamato:
Inhiben receptores del glutamato como el AMPA, kainato y NMDA, reduciendo la excitabilidad y aumentando el umbral convulsionante.
Apertura de los canales de potasio:
Se retarda la fase despolarizante por la apertura de los canales de potasio, esto estabiliza el potencial de membrana en reposo.
Inhibición de la proteína SV2A:
Se une a la Proteína 2A de la vesícula presináptica (SV2A) y la inhibe, proteína que parece estar involucrada en la fusión de las vesículas y la exocitosis de neurotransmisores.

También utilizados para dolores neuropáticos crónicos en vez de utilizar opiáceos
- http://www.infodoctor.org/dolor/CP077.html
- http://www.sedolor.es/index.php
- http://www.fibromialgia.nom.es/
- http://scielo.isciii.es/scielo.php
- http://www.medwave.cl
- La deficiencia de succínico semialdehído deshidrogenasa
Tabla I. Farmacoterapia anticonvulsiva según el síndrome epiléptico
Síndrome/Crisis 1a Elección 2a Elección 3a Elección
Parciales
- Sintomatología elemental Fenitoína Gabapentina
- Complejas (Psicomotoras) Carbamazepina
- Generalización secundaria Fenitoína Fenobarbital Gabapentina
Generalizadas
- Tónico clónicas (Gran Mal) Fenitoína Fenobarbital Carbamazepina
- Ausencias (Pequeño Mal) Ácido Valproico Etosuximida
- Espasmos infantiles Fenobarbital
Estatus Epiléptico Diazepam i.v. bolo Fenitoína
Diazepam inf. lenta
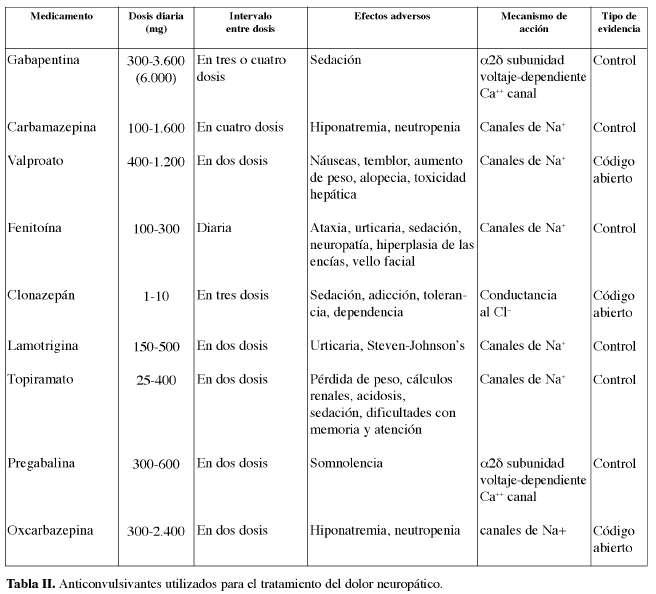
La gabapentina es eliminada sin metabolizar por el riñón, por lo que se debe modificar la dosis en caso de insuficiencia renal. No se han descrito interacciones significativas con otros antiepilépticos. El felbamato se ha asociado con anemia aplasica. Fue aprobado en 1993 para su uso en poli o monoterapia en adultos con crisis parciales con o sin generalización secundaria, y en niños con crisis parciales o generalizadas asociadas al síndrome de Lennox-Gastaut. Las principales interacciones farmacocinéticas entre los FAE son: FB, fenobarbital; PRM, primidona; DPH, fenitoina; ESM, etosuximida; CBZ, carbamacepina, VPA, ácido valproico; VGB, vigabatrina; GBP, gabapentina y LTG, lamotrigina.
 Farmacocinética y Farmacodinamia de los antiepilépticos
Farmacocinética y Farmacodinamia de los antiepilépticos
Farmacocinética de algunos antiepilépticos
Tabla FAE según el tipo de crisis
FB |
PRM |
DPH |
ESM |
CBZ |
VPA |
VGB |
GBP |
LTG |
Parciales |
+ + |
+ + |
+ + |
- |
+ + |
+ |
+ + |
+++ |
+ |
Generalizadas |
+ + |
+ + |
+ + |
- |
+ + |
+ + |
+ |
- |
+ |
Ausencias típicas |
- |
- |
- |
+ |
- |
+ + |
- |
- |
( + ) |
Ausencias Atípicas |
- |
- |
- |
- |
- |
+ |
- |
- |
+ |
Crisis atónicas |
- |
- |
- |
- |
- |
( + ) |
- |
- |
+ |
Crisis mioclónicas |
- |
- |
- |
- |
- |
+ + + |
- |
- |
( + ) |
Espasmos infantiles |
- |
- |
- |
- |
- |
+ + |
+ + |
- |
- |
Fuentes:
- http://bvs.sld.cu/revistas/ped/vol77_2_05/ped06205.htm
- http://emedicine.medscape.com/article/1187334-overview Con gráficos sencillos que explican como actúan los diferentes tipos de antiepilépticos
- http://www.saludmed.com/FisiolEj/NervoN.html
- http://www.uam.es/departamentos/medicina/anesnet/forconred/neuro/epilepsia/epilepsia1.htm
- http://www.todosobreepilepsia.com/
- http://bvs.sld.cu/revistas/ped/vol77_2_05/ped06205.htm
- http://escuela.med.puc.cl/publ/cuadernos/2004/EfectosAdversos.html
- http://www.apiceepilepsia.org
- http://www.epilepsyfoundation.org/
- http://farmafitolab.med.uchile.cl/download/Obstetricia/Anticonvulsivantes.doc
- http://neurologia.rediris.es/congreso-1/conferencias/epilepsia-6.html
- http://html.rincondelvago.com/epilepsia_5.html
- http://html.rincondelvago.com/antiepilepticos.html
- NUEVOS FARMÁCOS ANTIEPILÉPTICOS
Clasificación por estructura química:
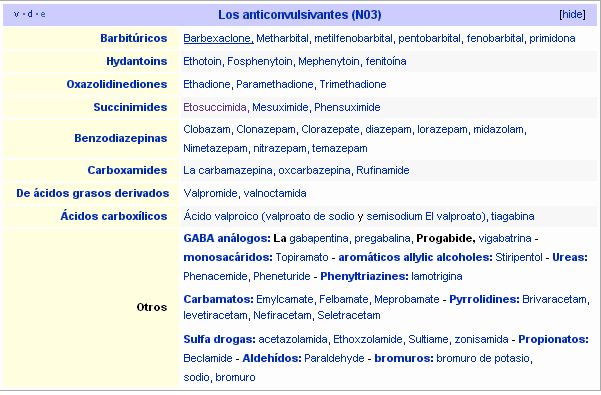
Fuente: Wikipedia
ANTICÍCLICOS (REGULADORES DEL ÁNIMO):
Metales:
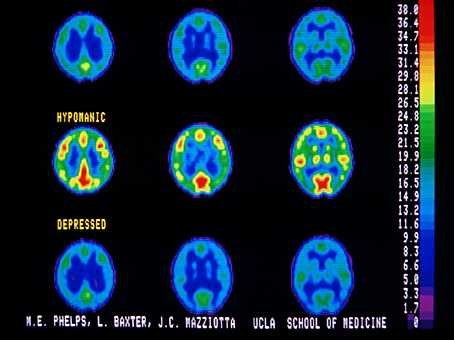
Programa de Investigación, Trastornos del Humor, encontraron que la amígdala parece ser ampliada significativamente en pacientes con enfermedad bipolar, junto con dilatación de los ventrículos.(Foto © UCLA).
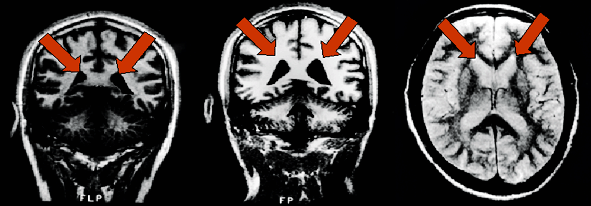
De izquierda a derecha: vista de un cerebro normal, el paciente con trastorno bipolar tiene dilatación de los ventrículos, manchas de color blanco brillante de hiperintensidad asociados con la enfermedad bipolar.
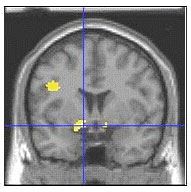
La amígdala izquierda (zona amarilla, donde las líneas se cruzan) y las estructuras relacionadas forman parte de un circuito cerebral que regula la emoción donde los niños con trastorno bipolar mostraron una mayor activación que los controles en la calificación de temor ante rostros neutrales. Estructura de la imagen con resonancia magnética funcional MRI datos superpuestos.
La amígdala es clave para nuestras experiencias emocionales. De esta estructura subcortical dependen los estímulos a los que respondemos, el modo en que están organizadas las respuestas que manifestamos a estos estímulos así como las respuestas internas de los órganos de nuestro cuerpo. Es como un gran integrador de aferencias sensitivas para modular respuestas efectoras en relación al medio externo e interno. En primates no humanos, tras lesión de la amígdala, los objetos que representaban una amenaza dejan de provocar miedo, o no se distinguen los objetos no comestibles de los comestibles. Por el contrario, la estimulación eléctrica de la amígdala intacta según el lugar concreto en el que se aplique evoca en los carnívoros diversas reacciones viscerales y de defensa. (Martin, 2001, p.457).
Fuentes:
- http://www.nih.gov/news/pr/may2006/nimh-29.htm
- http://es.wikipedia.org/wiki/Am%C3%ADgdala_cerebral
- http://mural.uv.es/locen/Diseno%20de%20trabajos%20cientificos.html(Importante)
Etiología
El trastorno bipolar no es ni fisiológico puro ni tampoco ambiental, es multifactorial, lo que significa que existen muchos factores genéticos y ambientales que, integrados, originan el trastorno.No hay una causa única para el trastorno bipolar—sino el acuerdo científico es que son muchos factores que actúan en conjunto y producen la enfermedad. Debido a que el trastorno bipolar tiende a prevalecer en las familias, los investigadores han tratado de buscar un gen específico que se transfiera por generaciones y el cual pueda incrementar las posibilidades de una persona de desarrollar la enfermedad. Con ello, la búsqueda mediante investigación de genes sugiere que el trastorno bipolar, como otras enfermedades mentales, no ocurre debido a un sólo gen.21
Los factores psicológicos también desempeñan un importante papel en la psicopatología del trastorno, así como en los factores psicoterapéuticos—cuyo objetivo es el alivio de los síntomas centrales—,el reconocimiento de los desencadenantes de episodios, el reconocimiento de los síntomas prodrómicos antes de una recurrencia declarada y la práctica de los factores que conducen a la continuidad en la remisión (Lam et al, 1999; Johnson & Leahy, 2004; Basco & Rush, 2005; Miklowitz & Goldstein, 1997; Frank, 2005). Las modernas psicoterapias basadas en la evidencia diseñadas específicamente para el trastorno bipolar, usadas en combinación con los tratamientos farmacológicos estándar aumentan el tiempo en que el individuo está bien significativamente más que con el uso exclusivo de medicación (Frank, 2005). Estas psicoterapias son la Terapia interpersonal y de ritmo social para el trastorno bipolar, la terapia familiar dirigida al trastorno bipolar, la psicoeducación, la terapia cognitiva para el trastorno bipolar y la detección prodrómica. Sin embargo, aún no se ha podido desarrollar un modelo cognitivo exhaustivo y general del trastorno bipolar.22
Eventos exógenos
Se ha relacionado anormalidades en la función cerebral a los sentimientos de ansiedad y una menor tolerancia al estrés. Cuando encaran un acontecimiento vital negativo de importancia, provocándoles éste un gran estrés, como un fracaso en un campo importante de sus actividades, pueden sufrir su primer episodio de depresión mayor. Por el contrario, cuando un individuo obtiene un gran logro puede experimentar su primer episodio maníaco o hipomaníaco. Los individuos con trastorno bipolar tienden a experimentar desencadenantes de episodio que suponen acontecimientos interpersonales o relacionados con logros personales. Ejemplos de sucesos vitales interpersonales serían el enamoramiento o por el contrario la muerte de un amigo íntimo. Entre los sucesos vitales relacionados con logros están la aceptación dentro de una élite o por el contrario, un despido (Miklowitz & Goldstein, 1997). Un nacimiento también puede desencadenar una psicosis postparto en las mujeres bipolares.
La "Teoría del kindling" (El kindling es un término que no se traduce. Significa astillas en inglés y se refiere a un aumento de excitabilidad en las neuronas del sistema límbico23 ) afirma que las personas que están genéticamente predispuestas al trastorno bipolar pueden experimentar una serie de acontecimientos estresantes, cada uno de los cuales disminuye el umbral al que puede darse un cambio de estado de ánimo.24 Eventualmente se puede desencadenar un episodio (que de este modo se hace recurrente) por sí mismo. No obstante, no todos los individuos experimentan a consecuencia de ello episodios en ausencia de acontecimientos vitales positivos o negativos.
Los individuos con una aparición del trastorno al final de la adolescencia o en la edad adulta temprana habrán experimentado con toda probabilidad ansiedad y depresión en la infancia. Existen argumentaciones en el sentido de que la aparición en la infancia del trastorno debería ser tratado cuanto antes.
Un historial familiar de trastornos dentro del espectro bipolar puede transmitir una predisposición o carga genética a desarrollar uno de estos trastornos.25 Puesto que los trastornos bipolares son poligénicos susceptibles de sufrir muchos trastornos bipolares y unipolares dentro del mismo pedigrí familiar. Este es frecuentemente el caso (Barondes, 1998). Los trastornos de Ansiedad, la depresión clínica, los trastornos de la alimentación, el trastorno disfórico premenstrual, la depresión postparto, la psicosis postparto y/o la esquizofrenia pueden ser parte del historial familiar, que es a lo que se refiere el término "carga genética".
Ya que el trastorno bipolar es tan heterogéneo, es probable que las personas que lo padecen experimenten distintas manifestaciones de la enfermedad (Miklowitz & Goldstein, 1997).
Recientes investigaciones realizadas en Japón señalan una hipótesis sobre un posible origen mitocondrial de este trastorno.(Stork & Renshaw, 2005).
Heredabilidad o herencia
El trastorno es prevalente según el historial familiar, es decir, que suele haber antecedentes familiares bien de trastorno bipolar o de otro tipo de trastorno del humor como la depresión.26 Más de dos tercios de las personas que padecen trastorno bipolar han tenido al menos un pariente cercano con el trastorno o con depresión mayor unipolar.
Cada vez existen más pruebas de un componente genético como causa del trastorno bipolar, proporcionado por algunos estudios en gemelos y de ligamiento genético. Los estudios que buscan la identificación de las bases genéticas del trastorno bipolar indican que la susceptibilidad procede de múltiples genes.27 Los investigadores tienen la esperanza de que identificando los genes de susceptibilidad y las proteínas que codifican se haga posible el desarrollo de mejores tratamientos e intervenciones preventivas que tengan como objetivo los procesos subyacentes de la enfermedad.
La tasa de concordancia genética en gemelos del trastorno es del 70%. Esto significa que si una persona tiene el trastorno, un gemelo idéntico tiene un 70% de probabilidad de padecer también el trastorno. Los mellizos tienen un 23% de tasa de concordancia. Estas tasas no se reproducen de manera universal en la literatura. Estudios recientes han observado una tasa de aproximadamente un 40% entre gemelos y menos de un 10% entre mellizos. (ver Kieseppa, 2004 and Cardno, 1999).28 29
En 2003 un grupo de investigadores de EEUU y Canadá publicaron un artículo que utilizaba técnicas de ligamiento genético para identificar una mutación en el gen GRK3 como posible causa de más del 10% de los casos de trastorno bipolar. Este gen corresponde a una enzima asociada a la quinasa, cuyo nombre es quinasa del receptor de la proteína G 3, que parece estar implicada en el metabolismo de la dopamina y puede proporcionar un posible objetivo para el diseño de nuevos fármacos para el trastorno bipolar.30
En 2007 un estudio de ligamiento genético ha identificado algunos genes que probablemente están implicados en la etiología del trastorno bipolar, lo cual sugiere que este trastorno puede ser una enfermedad poligénica. Los investigadores encontraron una correlación entre la DGKH (la diacilglicerol kinasa η) y el trastorno bipolar.17 Esta enzima es clave en la ruta del fosfatidil inositol sensible al litio.31
Patogenia
La fisiopatología y sus mecanismos subyacentes son pocos entendidos para el trastorno bipolar. La evidencia de los estudios preclínicos hasta ahora publicados sugieren que pueda compartir algunos mecanismos biológicos con la epilepsia. Se ha planteado que exista un desequilibrio entre aminoácidos excitadores, fundamentalmente glutamatos, y los inhibidores, principalmente el ácido γ-aminobutírico y la disfunción de las bombas de cationes como las bombas de sodio y calcio que explica la patogenia del trastorno bipolar y otras patologías como la epilepsia.2
Cuadro clínico
El trastorno bipolar es comúnmente encuadrado como Trastorno Bipolar Tipo I, en donde el individuo experimenta experiencias extremas de manía, o Trastorno Bipolar Tipo II, en donde los «altos» de la hipomanía no llegan hasta el extremo de la manía. Este último es mucho más difícil de diagnosticar, ya que los episodios de hipomanía pueden simplemente parecer como períodos de una alta productividad del individuo y se han reportado mucho menores que la depresión ansiosa. Puede ocurrir la psicosis, particularmente en los períodos de manía. También nos encontramos con subtipos de «ciclos acelerados». Debido a que en los problemas relacionados con los cambios de humor hay tantas variaciones en cuanto a su severidad y a su naturaleza, el concepto de espectro bipolar es usualmente utilizado, incluyendo en él la ciclotimia. No hay consenso en cuanto a la cantidad de «tipos» existentes de trastorno bipolar (Akiskal and Benazzi, 2006). Muchas personas con trastorno bipolar experimentan de una severa ansiedad y son muy irascibles cuando se encuentran en un período maniaco (hasta el punto de la furia), mientras que otros se vuelven eufóricos y grandilocuentes.
***
Actualmente no existe cura para el trastorno bipolar, pero puede ser controlado. El objetivo del tratamiento consiste en un control eficaz del curso de la enfermedad a largo plazo, lo cual puede suponer el tratamiento de los síntomas emergentes. Para lograrlo se emplean técnicas farmacológicas y psicológicas. Se emplean varios fámacos en el tratamiento, necesitándose en la mayor parte de los casos una combinación de varios. Sin embargo, existe poca evidencia médica de que el solo uso de tratamientos alternativos o complementarios pueda funcionar en el tratamiento a largo plazo.
Principios
Se usa un tipo de fármacos, llamados estabilizantes del estado de ánimo para prevenir o mitigar episodios de manía o depresivos. Entre los medicamentos de este tipo que han demostrado su eficacia están el litio y anticonvulsivantes como el ácido valproico, la carbamazepina y la lamotrigina. Todos los antipsicóticos atípicos están aprobados por la FDA para el tratamiento de estados agudos de manía. Los antipsicóticos. En términos generales, los estabilizadores del estado de ánimo son más eficaces en el tratamiento y la prevención de episiodios maníacos asociados con el trastorno bipolar. Sin embargo, otras medicaciones (p.ej. lamotrigina, fluoxetina, quetiapina) también han demostrado eficacia para el tratamiento de la depresión bipolar.
En medicina, toda medicación tiene efectos secundarios y la empleada en el trastorno bipolar no constituye una excepción. Es importante hacer notar que cada medicación está asociada a un perfil único de efectos secundarios.
El litio está vinculado con desarreglos gastrointestinales (nauseas, diarreas), problemas de memoria, ganancia de peso y otros. El aumento de la dosis corresponde con más efectos secundarios, pero dosis menores (dentro de la ventana terapéutica) tienen pocos o ningún efecto secundario.
Los anticonvulsivantes suelen provocar sedación, ganancia de peso, alteraciones de electrolitos y otros efectos secundarios. Si no se tolera bien un anticonvulsivo, es recomendable probar con otro. Una combinación de dos o más anticonvulsivos suele ofrecer resultados reduciendo la dosis eficaz de cada uno de ellos y disminuyendo los efectos secundarios.
El perfil de efectos secundarios de los antipsicóticos atípicos varía ampliamente entre agentes. En términos generales, el más común de estos efectos es la sedación y las alteraciones metabólicas (p.ej. ganancia de peso, dislipemia, hiperglucemia). También puede provocar efectos secundarios extrapiramidales e inquietud. Los Antipsicóticos atípicos también conllevan un riesgo de provocar discinesia tardía. No obstante, el riesgo con los antipsicóticos atípicos más recientes es mucho menor que el asociado con la generación anterior (p.ej con haloperidol). Se piensa que el riesgo de discinesia tardía está proporcionado a la duración del uso de neurolépticos (aumenta aproximadamente en un 5% anual en pacientes no ancianos tratados con los antiguos antipsicóticos). Pacientes y facultativos necesitan poner atención y vigilar los síntomas de estos efectos secundarios cuidadosamente para que se pueda reducir su dosis o cambiarlo por otra medicación antes de que el mal progrese. El facultativo, por supuesto, debería ser consultado antes de realizar cualquier cambio en la dosis.
Las medicaciones funcionan de diferente forma en cada persona y lleva un tiempo considerable determinar en cada caso en particular si un cierto fármaco es completamente eficaz y puesto que el trastorno bipolar es episódico por naturaleza y los pacientes pueden experimentar remisión tanto si reciben tratamiento como si no. Por esta razón, ni los pacientes ni los médicos tendrían que esperar un alivio inmediato, aunque la psicosis con manía puede responder rápidamente a antipsicóticos y la depresión bipolar se puede aliviar rápidamente con terapia electroconvulsiva. Muchos doctores hacen hincapié en que los pacientes no deberían una estabilización completa hasta después de al menos 3-4 semanas (algunos antidepresivos, por ejemplo, necesitan entre 4 y 6 semanas para hacer efecto) y no deberían abandonar la medicación prematuramente, ni deberían discontiar la medicación con la deaparición de los síntomas puesto que la depresión podría volver.
Uno de los principales problemas está en aceptar la medicación, puesto que algunas personas a medida que entran en manía pierden la percepción de estar enfermos y por ello discontinúan el tratamiento. Los pacientes también suelen cesar de tomar la medicación cuando los síntomas desaparecen, pensando erróneamente que están "curados", mientras que otros disfrutan con los efectos de la hipomanía no medicada.
La depresión no responde instantáneamente cuando se retoma la medicación y típicamente lleva más de 6 semanas la respuesta. La manía puede desaparecer lentamente o puede trasformarse en depresión.
Otras razones que refieren las personas para discontinuar la medicación son los efectos secundarios, el coste del tratamiento y el estigma de tener un trastorno psiquiátrico. En un número relativamente pequeño de casos estipulados por la ley (depende de lugares, pero suele estar en los ordenamientos jurídicos, solo cuando el paciente significa una amenaza para si mismo o para los demás), los pacientes que no están de acuerdo con su diagnóstico psiquiátrico y el tratamiento pueden ser forzados legalmente a recibir tratamiento sin su consentimiento.
Sales de litio
El uso de sales de litio como tratamiento el trastorno bipolar fue descubierto por el Dr. John Cade, un psiquiatra australiano, quien lo publico en un artículo sobre el uso del litio en 1949.
Las sales de litio han sido usadas desde hace mucho tiempo como tratamiento de primera línea en el tratamiento del trastorno bipolar. Antiguamente, los médicos enviaban a los pacientes con enfermedades mentales a beber de "fuentes alcalinas" como tratamiento. Aún sin saberlo estaban en realidad prescribiendo litio, que estaba presente en altas concentraciones en esas aguas. El efecto terapéutico parecía deberse por completo al ión de litio, Li+.
Las dos sales de litio que se usan en la terapia del trastorno bipolar son, principalmente el carbonato de litio y en otras ocasiones el carbonato de litio. Su uso fue aprobado por la FDA en 1970, siendo un estabilizante del estado de ánimo para muchas personas con este trastorno. También se ha comprobado que reduce el riesgo de suicidio.Aunque el litio es uno de los estabilizantes del estado de ánimo más eficaces, produce muchos efectos secundarios similares a los que experimentan los que ingieren demasiada sal de mesa, como elevación de la presión sanguínea, retención de agua y estreñimiento. Se precisan análisis de sangre regulares cuando se está tomando litio para determinar los niveles correctos ya que la dosis terapéutica está próxima a la dosis tóxica.
Se cree que el tratamiento por sales de litio funciona así:algunos síntomas del trastorno bipolar parece que se deben a la enzima inositol monofosfatasa (IMPasa), una enzima que escinde el inositol monofosfato en inositol libre y fosfato. Está implicado en la transducción de señales y se cree que crea un desiquilibrio en los neurotransmisores de los pacientes de trastorno bipolar. Se piensa que el ión litio produce un efecto estabilizante inhibiendo la IMPasa por sustitución de uno de los dos iones magnesio que están normalmente en el lugar activo de la IMPasa, disminuyendo su velocidad catalítica.
Ver: http://psicofarmacologia.info/estabilizadores.html
El orotato de litio se usa como tratamiento alternativo al carbonato de litio por algunos afectados, principalmente porque está disponible sin prescripción facultativa. Se vende en ocasiones como "litio orgánico" por nutricionistas, así como bajo muchas marcas comerciales. Parece que existen pocas pruebas en ensayos clínicos sobre su uso en comparación con el carbonato de litio. Los pacientes se quejan de que es mucho más débil que el carbonato de litio, y por tanto, menos eficaz.
Anticonvulsivos como estabilizantes del estado de ánimo
Los anticonvulsivos, en especial el valproato y la carbamazepina, se han usado como alternativas o coadyuvantes del litio en muchos casos. El valproato (Depakote, Depakene, Epival, etc...) fue aprobado por la FDA para el tratamiento de la manía aguda en 1995 y algunos médicos lo consideran ahora la terapia de primera elección en el trastorno bipolar. Para algunos es preferible al litio porque su perfil de efectos secundarios parece ser menos severo, la aceptación de la medicación es mejor y hay menos brotes de episodios maniacos. No obstante no es tan bueno como el litio en lal prevención de los episodios depresivos, de modo que los pacientes que toman valproato pueden necesitar la administración conjunta de antidepresivos como coadyuvantes.
Nuevos trabajos de investigación sugieren que diferentes combinaciones de litio y anticonvulsivos pueden ser útiles. estos últimos también se usan en combinación con antipsicóticos. Las nuevas medicaciones anticonvulsivas, como la lamotrigina y la oxcarbazepina son también eficaces como estabilizantes del estado de ánimo en el trastorno bipolar. La Lamotrigina es particularmente prometedora puesto que alivia la depresión bipolar y evita la recaída con altas tasas.
La Zonisamida (Zonegran), otro anticonvulsivo, parece ser un tratamiento prometedor según Frederick K. Goodwin M.D. en una reciente publicación en Medscape titulada "El diagnóstico correcto y el tratamiento a largo plazo de la depresión bipolar.
El Topiramato no ha tenido un buen comportamiento en los ensayos clíncos. Parece que le va muy bien a algunos pacientes pero a la mayoría no. Parece que es util en algunos casos resistentes al tratamiento y para problemas de ansiedad cuando no se puede prescribir clonazepam. Al ensayar Gabapentina, los pacientes no lograban distinguirla del placebo como estabilizante del ánimo.
De acuerdo con estudios realizados en Finlandia en pacientes con epilepsia, el valproato puede aumentar los niveles de testosterona en chicas adolescentes y producir síndrome de ovario poliquístico cuando comienzan a tomarlo antes de los 20 años. El aumento de testosterona puede producir este síndrome además de reglas ausentes o irregulares, obesidad y crecimiento anormal del pelo. Por tanto, las pacientes jóvenes que toman valproato deberían ser cuidadosamente observadas por el facultativo. No obstante, la dosis terapéutica para el paciente que toma valproato para una epilepsia es mucho mayor que la indicada para los que sufren trastorno bipolar.
Otros anticonvulsivos eficaces en algunos casos y que están siendo estudiados serían: fenitoína, levetiracetam, pregabalina y valnoctamida.
Fuente:
- http://es.wikipedia.org/wiki/Bipolar
- http://www.bipolarweb.com/
- http://www.bipolares.cl/
- http://www.mundobipolar.org/
- http://es.wikipedia.org/wiki/Manía
- http://es.wikipedia.org/wiki/Hipomanía
- http://www.psiquiatriabiologica.org.co/avances/vol7/doc/4_glutamato.pdf
COMENTARIOS DE: Gabapentina
LA GABAPENTINA PUEDE AUMENTARME DE PESO, ME PUEDE HACER OBESO?
Reacciones Adversas: Las más comunes son somnolencia, ataxia, fatiga, mareo y nistagmo. Con menor frecuencia se presentan: temblor, rinitis, ambliopía, diplopía, aumento de peso, faringitis, nerviosismo, disartria, amnesia, dispepsia, mialgias, dolor de espalda, depresión, tos, edema periférico, sequedad de boca y garganta, impotencia, alteraciones en el pensamiento, constipación, trastornos dentales, contracción espasmódica, erosión cutánea, prurito, vasodilatación, aumento del apetito, leucopenia, coordinación anormal y leucopenia.

la gabapentina como reaccion abversa puede causar disfuncion erèctil, entre otras reacciones. respuesta de Lupita si puede tomar sibutramina, no aconsejo ese medicamento si es eficaz para disminuir el apetito pero con mucha frecuencia causa sequedad bucal, estudios recientes afirman que lo suspendieron en Europa ya que aumenta el riesgo de sufrir Cardiopatias, lo mejor es comer saludablemente las tres comidas, eliminar refrescos, come bajo sal, bajo alimentos ricos en azucar, disminuir los alimentos grasos, consumir frutas, vegetales, pescados, mariscos, eliminar de la dieta comidas chatarras coo perro calientes, tacos,pepitos, hamburguesas. otro tips que funciona es comer despacio, amsticar bien los alimentos.
Estimados amigos: Tomo carbamazepina 1-1- 2 ( 800 mg al día) desde 1983; por lesion en lobulo temporal derecho que me provocaba depresiones cíclicas; una especie de epilepsi me explico mi Médico Psiquiatra. Ahora tengo 59 años estoy estable desde 2000; tengo mucho dolor en lumbares que baja las piernas, y a veces se me duermen las piernas, cuando el dolor es intenso; no tolero esta mucho de pie, ¿si tomo Gabapentina se contrapone con los 800 mg de carbamezepina que tomo diario?
Tuve un accidente en 1997, y tuve un fractura en lumbares y la cadera derecha se luxo y se salió de su lugar; me la acomodaron d e nuevo; pero me dijeron que probablemente despues se necrosara la cabeza del femur; tengo sobrepeso.
Gracias por su respuesta
María Guadalupe
¿si tomo Gabapentina se contrapone con los 800 mg de carbamezepina que tomo diario?
No, por farmacodinámica tienen diferente lugar de acción la gabapentina bloquea canales de calcio a2o dependientes de voltaje y la carbamazepina, bloquea canales de sodio y por farmacocinética no hay interacciones.

Tengo dolor neuropatico, y tomo gabapentina. aunque yo no tengo deabetes, yo quiero saber si si la perdida de equlibrio del oido izq. este asociado al dolor neuropatico. note que con la gabapentina no tuve dolor menstrual que eran insoportables, si sera por la gabapentina?
La gabapentina se da para neuropatías de cualquier causa, hay una neuropatía llamada diabética periférica por causa de diabetes que dice no padecer. Lo de la pérdida del equilibrio puede tener como causa una neuropatía pero no tiene que ser del oído interno. Lo del dolor menstrual, aliviado por la gabapentina, pues sí, sirve para cualquier dolor de tipo crónico y no agudo.

Los anticonvulsivantes suelen provocar sedación, ganancia de peso, alteraciones de electrolitos y otros efectos secundarios. Si no se tolera bien un anticonvulsivo, es recomendable probar con otro. Una combinación de dos o más anticonvulsivos suele ofrecer resultados reduciendo la dosis eficaz de cada uno de ellos y disminuyendo los efectos secundarios.
***
Aumento de peso del litio
Es un índice de la mejoría clínica del enfermo. Produce aumento del apetito y de la ingesta alimentaria - en especial hidrocarbonada- por causa que aún se desconocen. También produce una modificación del metabolismo hidrocarbonado, que provoca una curva de tolerancia a la glucosa de tipo diabético
***
¿Cómo Pueden los Medicamentos Anticonvulsivos Ayudarle a Perder Peso? Investigadores creen que algunas personas obesas podrían comer en exceso ya que experimentan impulsos eléctricos sin control en las áreas del cerebro involucradas con las sensaciones de placer. Cuando esto pasa, podría causar ansias incontrolables de cosas como comida, alcohol o nicotina. Las personas con epilepsia también tienen impulsos descontrolados en las células nerviosas de sus cerebros. Los medicamentos anticonvulsivos como el topiramato y la zonisamida interfieren con estos impulsos eléctricos sin control, que calman los impulsos eléctricos y previenen los ataques. Los doctores que tratan la epilepsia observaron que los pacientes que tomaban topiramato y zonisamida tendían a perder una cantidad significativa de peso (un promedio de 11% de peso corporal inicial). Investigadores han observado que los pacientes obesos a quienes se les dio zonisamida parecían sentirse llenos más pronto y tener ansia disminuida de dulces. Debido a este efecto, algunos investigadores creen que estos medicamentos también podrían amortiguar la actividad de las células nerviosas en los centros de placer del cerebro, de este modo aliviando las ansias de comida.
http://www.empowher.com/media/reference/nueva-terapia-de-combinacion-para-controlar-la-obesidad
***
Pruebe con topiramato, parece que hay anticonvulsivos que hacen ganar peso a algunas personas y otros les adelgaza

HOLA SOY BIPOLAR Y LA SIQUIATRA M MEDICA CON LAMOTRIGINA 200 UNA DOSISI POR DIA Y 3 DOSIS DE CARBONATO DE LITIO 300 mg. HACE UN AÑO Q LO TOMO Y AUMENTE DE PESO Y NO PUEDO BAJAR QUISIERA SABER SI ES CAUSA DEL MEDICAMENTO Q ENGORDO Y SI HAY ALGO Q LO CONTRARESTE PARA PODER BAJAR SI ES QUE TOMANDO ESTOS REMEDIOS SE PUEDE BAJAR...MUCHAS GRACIAS

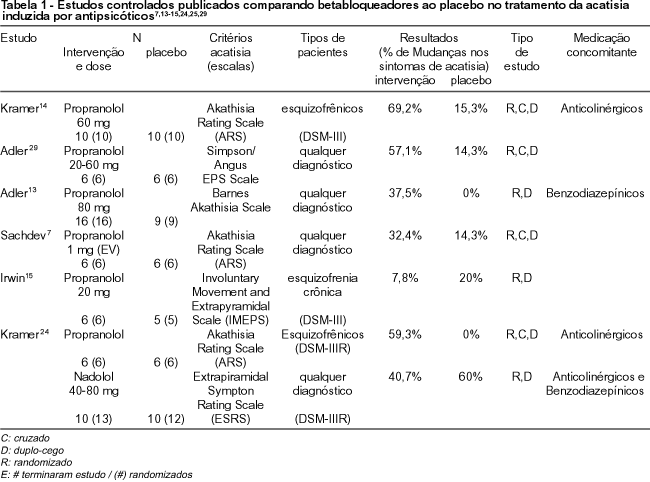{kind=link}
Le dejo un enlace
Disfunción sexual asociada al uso de gabapentina en el tratamiento del dolor central
|

|
Comentarios 1 a 10 de 17
6 meses para responder posts antiguos
¡Queremos tu opinión!
Si pregunta sobre este fármaco revise antes estos dos enlaces en particular
Ahora ya puede entender el prospecto y la ficha técnica

