CLASIFICACIÓN DE ANTIPSICÓTICOS
Nombre: Quetiapina
Nombre Comercial: Seroquel, Quetidin, Asicot, Quetiazic, Rocoz, Catepsin, Gofyl, Quetiamylan
Foto:
Fórmula: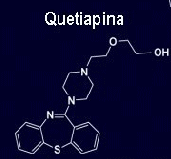
Información: http://es.wikipedia.org/wiki/Quetiapina
http://www.eutimia.com/psicofarmacos/antipsicoticos/quetiapina.htm
http://www.farmaciasahumada.cl/__/P5523.HTM
http://www.cochrane.org/__/ab000967.html
Fármaco consultado 242477 veces
Antipsicóticos Típicos:
Farmacodinamia También llamados neurolépticos, constituyen un grupo de medicamentos de naturaleza química muy heterogénea pero con mecanismo de acción común. Actúan fundamentalmente por bloqueo de los receptores dopaminérgicos D2, aunque muchos antipsicóticos tienen actividad sobre los receptores de otros neurotrasmisores.
En la actualidad se sabe que la eficacia antipsicótica está estrechamente relacionada con la acción antidopaminérgica a nivel de las vías córtico-meso-límbicas, por bloqueo de los receptores postsinápticos. (4) A su vez la mayoría de efectos adversos neurológicos y endocrinológicos dependen del bloqueo dopaminérgico, aunque no podemos ignorar el protagonismo de diferentes receptores en otros efectos secundarios (sedación por H1 y alfa1, hipotensión ortostática por alfa1, etc.).
Típicos Depot:
Biodisponibilidad de larga duración e inyectados por vía intramuscular, en su mayoría se forman por esterificación de su grupo hidroxilo con un ácido graso de cadena larga; los ésteres se disuelven en distintos tipos de aceite vegetal. Peligrosos por su lenta absorción como su eliminación por el organismo ya que sus metabolitos duran días en el organismo.
Enlace: http://www.imedicinas.com/GPTage/Open.php?Y2EwNHNlMDJzYjAy
Fenotiazinas:
Se caracterizan por una estructura de 3 anillos (anillos resultantes de la unión de dos anillos benzénicos a través de un puente de N y S. En el grupo de tioxantenos, el puente de N se reemplaza por un puente de C) y difieren entre ellas en las sustituciones realizadas en la posición 2 y 10 (R1 y R2) de la estructura química . En los neurolépticos, derivados fenotiazínicos, la cadena lateral en R1 posee siempre 3 átomos de C seguidos de 1 átomo de N. Esta cadena lateral, es indispensable para el mantenimiento de las propiedades antipsicóticas. La adición de un cloro en R2 origina una asimetría en el núcleo fenotiazínico, con lo que se incrementa la acción farmacológica. El agregado de un radical CF3 en la misma posición, incrementa aún más las acciones antipsicóticas y antieméticas de las fenotiazinas.
Derivados Alifáticos:
Estas fenotiazinas, tienen dos grupos metilos en el nitrógeno terminal de la cadena lateral (R1) . Poseen una acción sedativa evidente Principalmente antihistamínicos H1. Sus efectos tranquilizantes son intensos por lo que son utilizados comúnmente en episodios esquizofrénicos agudos, excitación maníaca, delirios, agitación ansiosa, etc. Cadena lateral Alifática baja potencia (menor efecto sobre receptores D2), pocos efectos extrapiramidales (3.5%) en comparación con los antipsicóticos convencionales incisivos, producen sedación e hipotensión.
Derivados Piperidínicos:
Son las drogas menos potentes contienen un grupo piperidina, en la cadena lateral. Principalmente antihistamínicos H1. La más conocida es la tioridazina, que tiene una indicación en casos de esquizofrenia con síndromes depresivos . Tomando como base las acciones de la clorpromazina, se ha demostrado que la flufenazina, es a dosis iguales, aproximadamente 20 veces más potente que aquella, y que la trifluoperazina y tioproperazina lo son 10 veces más. La tioridazina, por otra parte, posee solo la mitad de la actividad farmacológica de la clorpromazina. Antipsicóticos de mediana potencia con muy pocos efectos extrapiramidales (0.6%) y con acción sobre receptores 5-HT2
Piperidínicos Depot:
Lo dicho para los típicos depot.
Derivados Piperazínicos:
Son las fenotiazinas más potentes con un grupo piperazina o piperazinil en la cadena lateral. Su acción antipsicótica permite su uso crónico en pacientes esquizofrénicos. Prácticamente no provocan hipotensión ortostática, o su acción es muy pequeña, en este sentido. Son marcadas sin embargo sus acciones extrapiramidales (de mayor intensidad que las dimetílicas). Son activas en dosis menores que las CPZ, y su acción es más rápida provocando escasa acción sedativa. Fármacos más incisivos, con mayores efectos extrapiramidales (7%) y alto riesgo de discinesia tardía. Ventana terapéutica alta. Tienen acción sobre vías nigro-estriales mayor que sobre vías mesolímbicas. Con pocos efectos antimuscarínicos y autonómicos
Piperazínicos Depot:
Lo mismo que lo dicho en los típicos depot.
Tioxantenos:
Derivan del reemplazo del puente de N de las fenotiazinas por un puente de C , manteniéndose la cadena lateral correspondiente. El isómero cis tiene una mayor potencia que el isómero trans. Fármacos de extrapiramidalismo medio (4.1%), con efecto hipotensor, anticolinérgico, sedativo y antiserotoninérgico.
Butirofenonas:
Son compuestos sintéticos. El núcleo butirofenona es una cadena de tres átomos de C unido a un grupo cetónico, y a un anillo bencénico. Todas las butirofenonas, tienen también un átomo de flúor en posición para, del anillo bencénico y la cadena alifática se une a un nitrógeno terciario de un anillo de piperidina (similar a las fenotiazinas). Son potentes agentes neurolépticos de amplio uso psiquiátrico
4-ANILINOPIPERIDÍNICAS:
Caracterizadas por un anillo fenil sustituido. Medicamentos de alta potencia, con mayor efecto extrapiramidal (16%) y ventana terapéutica alta, pero con pocos efectos anticolinérgicos y menor riesgo ocular, hepático y hematológico
Ver gráfico: Correlación entre las dosis clínicamente eficaces de neurolépticos y su capacidad de desplazar al H-haloperidol de su fijación al estriado. (Según Seeman, con autorización de Williamns & Wilkins.)
Difenilbutilpiperidinas:
Tienen una gran liposolubilidad y lipofilia, por eso, los lípidos del organismo actúan como depósito de estas drogas, con escasa excreción y larga duración. Están relacionados químicamente con las butirofenonas, con una cadena de tres átomos de C, un anillo piperazínico, y una estructura de dos anillos bencénicos, con un átomo de flúor cada uno en el otro extremo de la cadena
Potentes; causan poca sedación y ninguna hipotensión, pero si un extrapiramidalismo medio
Análogos e isómeros de Fenotiazinas:
Tienen una estructura de tres anillos.
Dibenzoxazepinas:
La posición central lo ocupa la oxazepina, en la estructura tricíclica o heterocíclica
Enlaces:
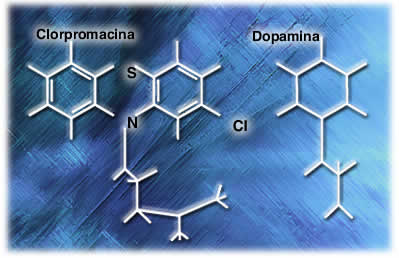
Similitud de estructura química ente la clorpromazina y la dopamina En 1964 los investigadores se dedicaron a buscar una sustancia endógena, químicamente similar a clorpromazina y encontraron dopamina, que aunque tiene una estructura química diferente, sus semejanzas pueden hacerlos activar un mismo receptor. Estudios posteriores demostraron que la clorpromazina se unía a los receptores de dopamina, pero por sus diferencias estructurales no era capaz de activarlo, actuando como antagonista
Enlaces:
Antipsicóticos Atípicos: Quetiapina
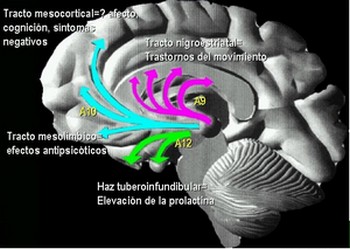
Farmacodinamia Meltzer & Nash clasificaron los antipsicóticos de acuerdo a su afinidad por receptores 5-HT2A v.s. D2, llamando a los que tenían actividad antiserotoninérgica, antipsicóticos atípicos (Meltzer, 1991). Sin embargo, muchos autores han cuestionado que las propiedades atípicas de estos antipsicóticos se deban al bloqueo serotoninérgico, y en cambio, han propuesto que la acción diferencial sobre vías dopaminérgicas A9 (nigroestriatales) y A10 (mesolímbicas) y la afinidad por el receptor D2 sean los responsables de las mismas (Gerlach, 1991 ; Corbett et al., 1993; Benkert & Wetzel, 1994; Vanelle et al., 1994; Seeman et al., 2000). Esto lo hacen partiendo de la observación de que la Risperidona, el cual también ha sido considerado un antipsicótico atípico, a dosis mayores de 6 mg./día produce síntomas extrapiramidales en un 30% de los pacientes, los cuales se van incrementando progresivamente con la elevación de las dosis hasta alcanzar un 50% con 16 mg./día, sugiriendo que el bloqueo de receptores 5-HT2 tiene una ventana protectora muy pequeña. Además, la Ritanserina, un antagonista de receptores 5-HT2 no logra antagonizar la catalepsia en ratas causada por el bloqueo de receptores D2 con Racloprida (Seeman, 1995).
Actualmente se considera que un antipsicótico atípico debe exhibir las siguientes propiedades: 1) disminuir los síntomas psicóticos, 2) disminuir los síntomas negativos, 3) disminuir los déficits neurocognitivos, 4) ser eficaz en pacientes refractarios, 5) pocos efectos extrapiramidales, 6) poca incidencia de discinesia tardía, 7) sin efectos en los niveles de prolactina
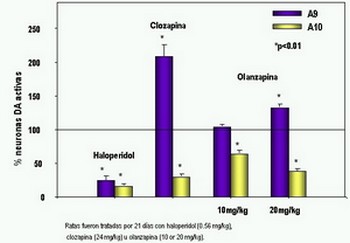
Estudios in vitro han demostrado que el predictor más importante de atipicidad de un antipsicótico es la disociación rápida de los receptores D2, teniendo mayor impacto que la alta afinidad a receptores 5-HT2.
En condiciones basales 25% a 40% de los receptores D2 están ocupados por dopamina. Los bloqueadores D2 compiten por estos sitios de unión. Entre más rápida sea la disociación del fármaco y el receptor, más rápida es la respuesta del medicamento a las elevaciones de niveles de dopamina. Clozapina tiene una velocidad de disociación 100 veces mayor que haloperidol.
Fuente: http://cuentospintados.com/portfolio/geodon/centro_enf/art3c.htm
Atípicos Depot:
Preparados depot de los atípicos en forma o no de estéres (esterificación de su grupo hidroxilo con un ácido graso de cadena larga), administrados por vía intramuscular, peligrosos por su lenta absorción y eliminación del organismo. Lo mismo dicho que en los típicos depot.
Benzamidas Sustituidas:
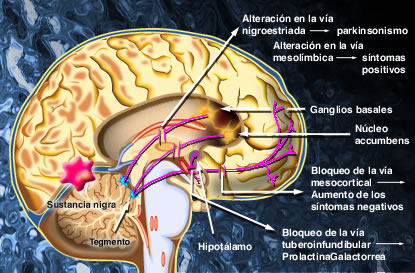
También llamados Ortopramidas, poco efecto extrapiramidal, efecto antiD2 a dosis muy elevadas, tienen mayor efecto de hiperprolactinemia: Por antagonismo dopaminérgico D2 a nivel tuberoinfundibular, se "libera" la inhibición dopaminérgica de la prolactina. Se presenta galactorrea, ginecomastia y amenorrea. Son enantiómeros de estos compuestos la metoclopramida (Primperan®, Reliveran®) y la cisaprida.
Derivados Amino-Etil:
Derivados. 2-Pirrolidinil:
Análogos e Isómeros de Fenotiazinas
Son también compuestos tricíclicos, en los que se reemplaza el puente de S de las fenotiazinas por un puente de N = C y una cadena lateral cíclica. Tienen la ventaja de desarrollar muy escasos efectos extrapiramidales, mínimas reacciones distónicas y parkinsonianas, aunque pueden ocurrir otros efectos adversos: como fatiga, mareos, o hipertensión, taquicardia y convulsiones, que tienden a desaparecer lentamente con el uso. A veces se observa una sialorrea o salivación intensa. Menor acción antagonista dopaminérgica D2, bloqueante serotoninérgico 5-HT2A y dopaminérgico D4.
Dibenzodiazepinas:
De estructura similar a los antidepresivos tricíclicos
(pero el anillo central posee 7 miembros), con una sustitución piperazínica en el anillo central.
Dibenzotiazepinas: Quetiapina
Al igual que los derivados benzisotiazólicos, presentan
menor afinidad para el bloqueo de los receptores 5-HT2A que el resto de los antipsicóticos atípicos. Sin embargo, conservan la relativa menor afinidad para el bloqueo de los receptores D2.
Dibenzoxepinas:
Pierden el nitrógeno, de la estructura tricíclica o heterocíclica central. Mayor bloqueo de los receptores 5HT que los de la dopamina
Tienobenzodiazepinas:
Compuestos que se asemejan químicamente a la clozapina, compartiendo alta afinidad para el bloqueo de los receptores 5-HT2A y una menor afinidad por los receptores D2.
Benzisoxazol:
Contiene una estructura benzisoxazol + piperidina. Combinan alta afinidad por los receptores dopaminérgicos D2 y por los receptores serotonérgicos 5-HT2A en dosis bajas, no tienen casi efectos sobre los receptores muscarínicos
Benzisotiazol:
Presentando una estructura de benzisotiazol + piperazina. Alta afinidad para el bloqueo de los receptores 5-HT2A y moderada a baja afinidad para el bloqueo de los receptores D2, H1y alfa1 adrenérgicos.
Indoles
Derivados Indólicos:
Tienen el núcleo indólico
La introducción de un metilo en el nitrógeno indólico supone un incremento notable de sus propiedades antiserotonínicas.
Derivan su efecto inhibidor selectivo sobre las neuronas dopaminérgicas mesolímbicas y es debido a que ejerce efectos inhibidores equilibrados sobre los receptores centrales D2 de la dopamina y 5HT2 de la serotonina, así como sobre los receptores a1 adrenérgicos. Tienen mucha menos afinidad para los receptores D2 que la mayoría de los agentes antipsicóticos y tiene relativamente baja afinidad para los receptores D1. Tiene sólo de baja a moderada afinidad por los receptores colinérgicos y alfa-adrenérgicos e histamínicos H1
2- Imidazolidinona:
Alcaloides de la Rauwolfia:
Los alcaloides (de álcali y -oide) son compuestos orgánicos nitrogenados de carácter alcalino producidos casi exclusivamente por vegetales (aunque también los hay producidos por animales y hongos, y otros sintetizados químicamente). Normalmente derivan de los aminoácidos.
Agonistas Parciales:
En farmacología, los agonistas parciales son fármacos que se unen y activan un dado receptor, pero que tienen sólo una eficacia parcial en el receptor con respecto a un agonista completo. También es posible considerarlos ligandos que muestran tanto efectos agonistas como antagonistas: cuando un agonista completo y un agonista parcial están presentes al mismo tiempo, el agonista parcial actúa como un antagonista competitivo, compitiendo con el agonista completo por la ocupación del receptor y produciendo una disminución neta en la activación del receptor observado con el agonista completo solo. Clínicamente los agonistas parciales pueden utilizarse para activar receptores para producir una respuesta submáxima deseada cuando no hay cantidades endógenas adecuadas del ligando, o pueden reducir la sobreestimulación de los receptores cuando hay cantidades excesivas del ligando.
Quinolinonas:
Derivado de la estructura de la quinolinona. Agonismo parcial sobre los receptores D2 y 5-HT1A y antagonismo 5-HT2A
Azapironas:
Derivado de la estructura de la azapirona. Agonismo parcial sobre el receptor 5- HT1A y antagonismo 5 HT2A. Ver también ansiolíticos, azapironas
Superatípicos: Fármacos que actúan sobre otros receptores que no sean dopoaminérgicos, como el sistema muscarínico colinérgico u otras inhibiciones.
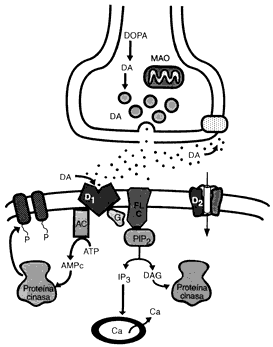
CICLO SINAPSIS DE LA DOPAMINA
Durante la actividad neuronal, la Dopamina es liberada de sus vesículas de almacenamiento. La cantidad almacenada y liberada depende de la capacidad disponible almacenada, de la proporción en que las vesículas son descargadas y recargadas y de la proporción en que nuevas vesículas son formadas.El incremento en la densidad de los receptores D2 siguiendo la administración crónica de antagonistas podría ser responsable del desarrollo de un desorden del movimiento denominado discinesia tardía.
Los agonistas a los receptores de dopamina, incluyendo las anfetaminas, bromocriptinas. Existe una fuerte correlación entre las dosis clínicas de los neurolépticos y su afinidad por los receptores D2 en el cerebro. Esto ha conducido a la hipótesis de que los desórdenes psicóticos son el resultado de una hiperestimulación de los receptores D2. La adminstración prolongada de neurolépticos a humanos o a animales de laboratorio pueden llevar a un incremento en la densidad de los receptores D2 del estriado y en la aparición de efectos secundarios extrapiramidales, incluyendo desórdenes del movimiento parkinsonianos y discinesia tardía. Las afinidades relativas de los receptores D2, D3 y D4 para los neurolépticos típicos y atípicos, conjuntamente con la expresión selectiva del ARNm para el receptor D3 en áreas límbicas del cerebro, ha conducido a la hipótesis de que la utilidad clínica de los neurolépticos en el tratamiento de enfermedades psiquiátricas puede ser debido, por lo menos en parte, a su capacidad para antagonizar la estimulación de los receptores D3 o D4, mientra que la disfunción motora observada al seguir un tratamiento crónico con neurolépticos típicos, podría ser debida a alteraciones en la densidad de los receptores D2 en el estriado.
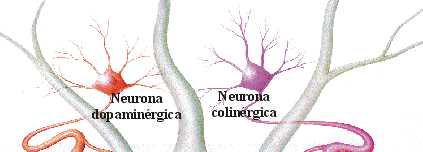
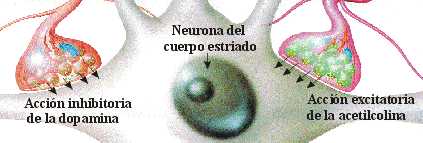
EQUILIBRIO DOPAMINA/ACETILCOLINA
Desde el núcleo caudado y el putamen, existe una vía hacia la sustancia negra que segrega el neurotransmisor inhibitorio GABA (ácido gamma aminobutírico). A su vez, una serie de fibras originada en la sustancia negra envía axones al caudado y al putamen, segregando un neurotransmisor inhibitorio en sus terminaciones, la dopamina. Esta vía mutua mantiene cierto grado de inhibición de las dos áreas y su lesión provoca una serie de síndromes neurológicos, entre los que se encuentra la enfermedad de Parkinson. Las fibras provenientes de la corteza cerebral segregan acetilcolina, neurotransmisor excitatorio, sobre el neoestriado. Las causas de las actividades motoras anormales que componen la enfermedad de Parkinson se relacionan con la pérdida de la secreción de dopamina por las terminaciones nerviosas de la sustancia negra sobre el neoestriado (tracto nigroestriatal) al que deja de inhibirlo. De esta forma, predominan las neuronas que segregan acetilcolina, emitiendo señales excitatorias a todos los núcleos de la base, responsables en conjunto, del planeamiento motor y algunas funciones cognitivas.
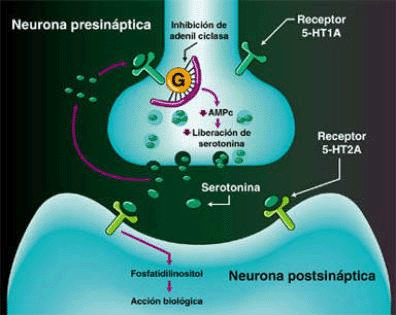
SEROTONINA
La clásica terapéutica antipsicótica se basa en el bloqueo dopaminérgico (D2) logrado con los neurolépticos tradicionales, con evidente acción sobre los síntomas positivos, pero escasa o nula sobre los negativos. A partir de la aparición de la Clozapina, primer antipsicótico atípico con acción predominantemente antagónica sobre los receptores 5HT2, se abre una nueva perspectiva en el tratamiento de la esquizofrenia. Se logra un efecto resocializante, antiagresivo, antiautístico, sin dejar de lado el efecto antidelirante y alucinolítico, y con escasa producción de efectos extrapiramidales. Este fármaco bloquea los receptores D2 mesolímbicos sólo en un 20%, mientras que lo hace en el 90% de los 5HT2.
En los cuadros psicóticos en los cuales predomina el componente disperceptivo, tienen su indicación específica los antagonistas 5HT3 (Ondansetrón, Granisetrón y otros). Añaden a su efecto alucinolítico una acción antidepresiva leve, y una acción neuroprotectora sobre la esfera afectiva y cognitiva.Receptores para la serotonina
No existe un receptor único para Serotonina, sino mas bien ha sido descrita toda una superfamilia de receptores con funciones específicas en las áreas pre y postsinápticas. Estudios farmacológicos y fisiológicos han contribuido a la definición de muchos subtipos de receptores para serotonina. Inicialmente se diferenciaron dos receptores diferentes de 5-HT en el íleon, llamados receptores D (bloqueado por dibencilina) y M (bloqueado por morfina). El receptor D se pensó que estaba en el músculo liso del íleon mientras que el receptor M, se consideró que estaba en la estructura ganglionar.
El desarrollo del ensayo de unión al radioligando fue propuesto por Pertoutka y Snyder en 1979 para etiquetar dos clases de receptores serotoninérgicos en el cerebro. Los lugares de unión con alta afinidad por [3H]-5-HT fueron designados como receptor 5-HT1; los lugares de unión etiquetados con alta afinidad por [3H]espiperona fueron denominados como receptor 5-HT2.
Las neuronas serotoninérgicas, originarias de los núcleos del rafe, ejercen un tono inhibidor sobre la transmisión dopaminérgica en las zonas nigroestriatales y mesocorticales limitando su síntesis y liberación. En la esquizofrenia esta inhibición de la dopamina por el control serotoninérgico está exagerada, lo que explica en parte la hipoactividad dopaminérgica nigroestriatal y mesocortical Esta inhibición puede ser levantada por las moléculas antagonistas serotoninérgicas.
La principal razón de la superioridad de los nuevos antipsicóticos con respecto a los neurolépticos convencionales es su propiedad antagonista 5-HT2 . El antagonismo de la actividad serotoninérgica en las zonas nigroestriatales y frontales permite una disminución de los síntomas deficitarios y una disminución de los efectos extrapiramidales, conduciendo a una mejor tolerancia.
Enlaces:
Dopamina |
Serotonina |
Noradrenalina o (Norepinefrina) |
Histamina |
 |
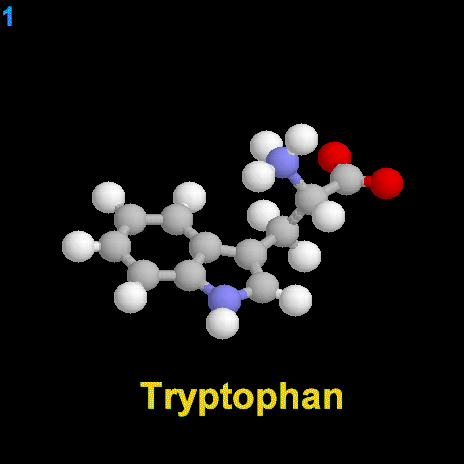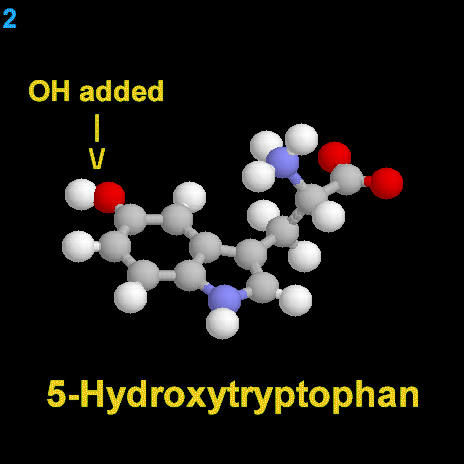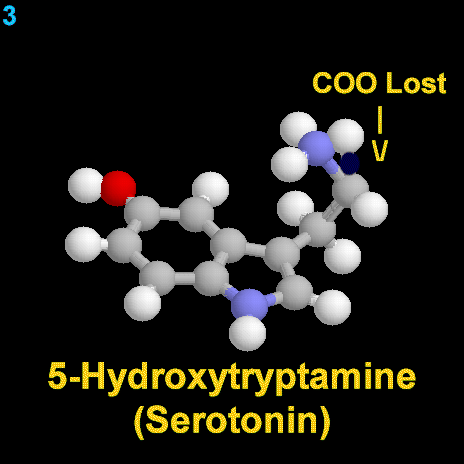 |
 |
 |
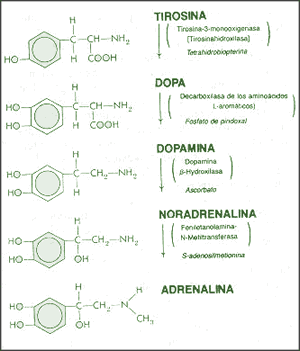

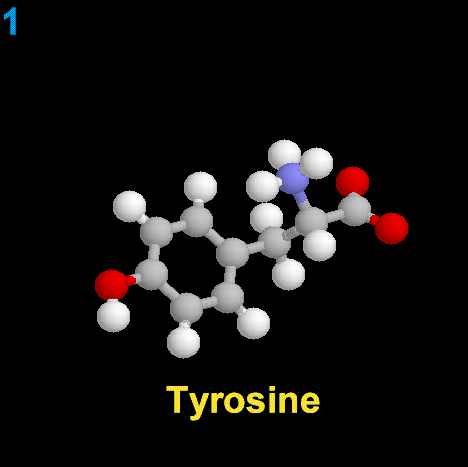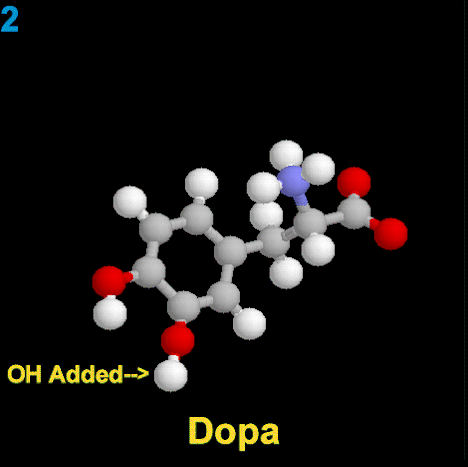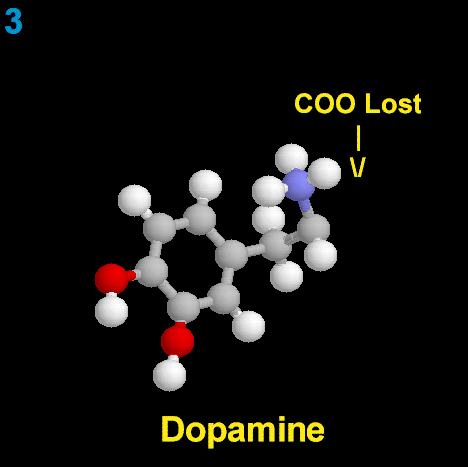
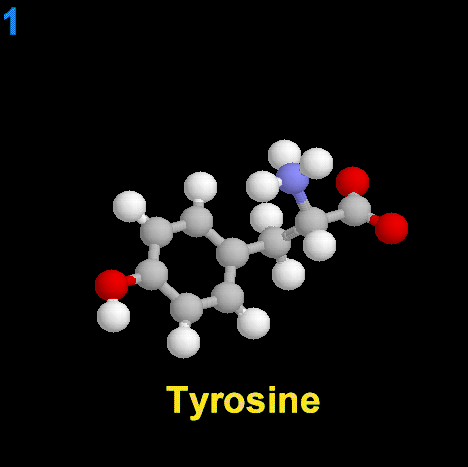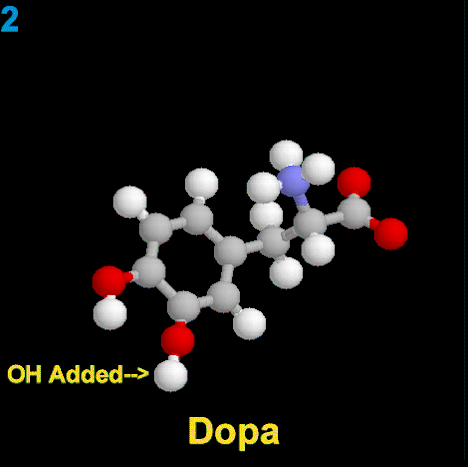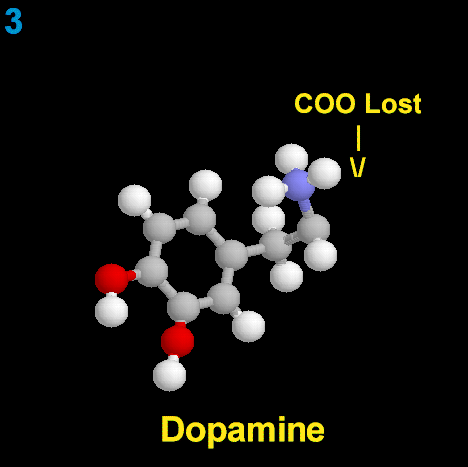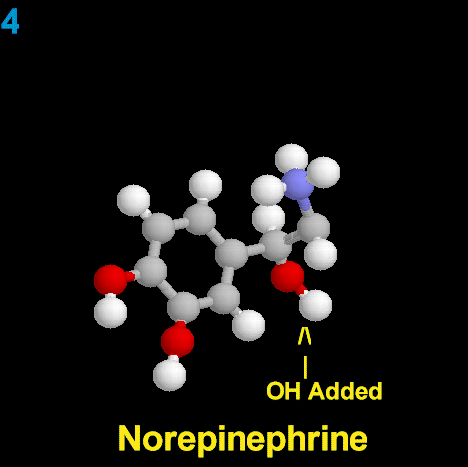
La dopamina es una monoamina del grupo de las catecolaminas, pues además del grupo amino solitario (-NH2), característico de las monoaminas, posee un grupo catecol, conformado por un anillo bencénico y 2 grupos hidroxilo (-OH).
La Dopamina es el precursor metabólico inmediato de la Noradrenalina y Adrenalina, sintetizada a partir de Tirosina que, por acción de la enzima Tirosinhidroxilasa es hidroxilada para convertirse en DOPA, que posteriormente es descaboxilada por la DOPA-descarboxilasa para dar Dopamina. Debido a su posición central en el metabolismo de las catecolaminas puede encontrarse Dopamina en cualquier lugar que se produzca Adrenalina o Noradrenalina. No obstante, su concentración a nivel del núcleo nigroestriado es mucho mayor que la de Noradrenalina. También hay neuronas dopaminérgicas en la retina, pero su función hasta la fecha es desconocida.
La biosíntesis de la dopamina está intrínsecamente relacionada con la NA, aunque su degradación está sujeta a los mismos sistemas enzimáticos, difiere en función de cual sea el primer sistema enzimático que actúe. Si actúa la monoaminooxidasa (MAO) se produce el ácido 3,4 dihidroxifenilacetico (DOPAC), que es un metabolito final de la dopamina; si actúa la COMT, hay un metabolito intermediario, la 3-metoxitiramina, momento en el cual interviene la MAO produciendo el producto final de la degradación, el ácido homovanílico (AHV).
La liberación es similar a la de la NA, siendo las anfetaminas unos potentes liberadores. Son inhibidores los a-hidroxibutiratos y la reserpina, que es también un depleccionante de la DA al igual que ocurría con la NA.
Por otra parte, la recaptura no debe seguir el mismo camino que la NA, puesto que los antidepresivos tricíclicos no la afectan y sin embargo, sí la anfetamina y la benzotropina.
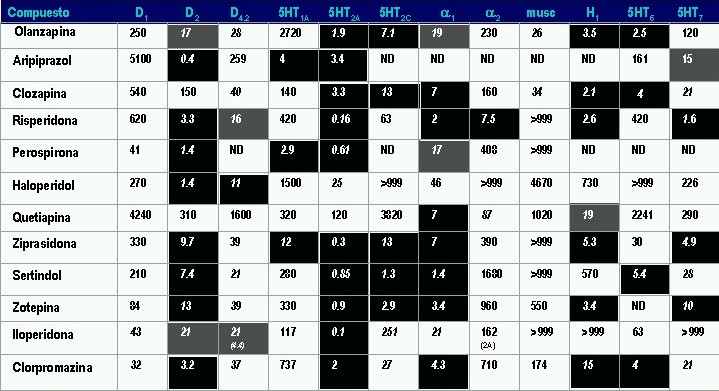
Las afinidades de los APs por diferentes receptores según su constante de inhibición (Ki) expresada en nanomoles se presenta en la siguiente tabla. En fondo negro se aprecian los receptores sobre los que cada uno de estos medicamentos presenta mayor afinidad (menor Ki) y en gris los que presentan moderada afinidad
Afinidad Receptores
Según constante de inhibición (Ki) expresada en nanomoles
Para comparar de forma gráfica la afinidad de diferentes antipsicóticos ir a --> búsqueda taxonómica de esta web.
Nombre: Quetiapina |
Gráfico y Leyenda:

|
| D1 |
D2 |
D3 |
D4 |
5-HT1A |
5-HT2A |
5-HT2B |
5-HT2C |
a1 |
a2 |
M1-5 |
H1 |
Dopamina D1 |
Dopamina D5 |
Dopamina D2 |
Dopamina D3 |
Dopamina D4 |
4240 |
|
160 |
|
1600 |
Serotonina 5-HT1A |
Serotonina 5-HT2A |
Serotonina 5-HT2B |
Serotonina 5-HT2C |
Serotonina 5-HT6 |
Serotonina 5-HT7 |
|
220 |
|
|
|
|
Noradrenalina a1 |
Noradrenalina a2 |
*Acetilcolina M1-5 |
Histamina H1 |
7 |
87 |
1020 |
11 |
Cuanto < más bajo es el valor, más > afinidad del receptor hay. Los valores difieren según la fuente farmacológica aquí son mayormente de Jesús Flórez completados con la tabla de arriba.
Antagonismo D2 = efecto sobre síntomas positivos [todos los antipsicóticos]; Agonismo 5-HT1A: efecto deletéreo en cognición [Aripiprazol]; Antagonismo 5HT2A: efecto favorable en cognición por normalización del funcionamiento NMDA [todos los atípicos y clorpormazina]; Antagonismo 5-HT2C: aumento del apetito [clozapina, olanzapina]; Antagonismo alfa 1: hipotensión, efecto sedante y mejoría de memoria de trabajo bajo estados de estrés [todos los antipsicóticos atípicos]; Antagonismo alfa 2: hipertensión [risperidona]; aumenta la liberación de noradrenalina; Efecto anticolinérgico central: compromiso de funciones cognoscitivas [clozapina]; Antagonismo H1: sedación, aumento de apetito [atípicos y clorpromazina]; Antagonismo 5-HT6: elevación de acetilcolina en espacio sináptico en corteza prefrontal e hipocampo (mejoría cognoscitiva)[olanzapina y clozapina] . En los receptores de acetilcolina, la mayor parte de fuentes hacen una media de los 5 receptores acetilcolinérgicos del tipo M1 (M1,M4,M5) activadores de la fosfolipasa C (Gq) y los del tipo M2 (M2,M4) inhibidores de la adenilato ciclasa (Gi), acciones contrapuestas pero dan el dato medio.
Fuente: psicofarmacologia.info (modificado)
Ver gráfico afinidad a receptores de algunos antipsicóticos atípicos
Enlaces:
Velocidad de disociación de los antipsicóticos
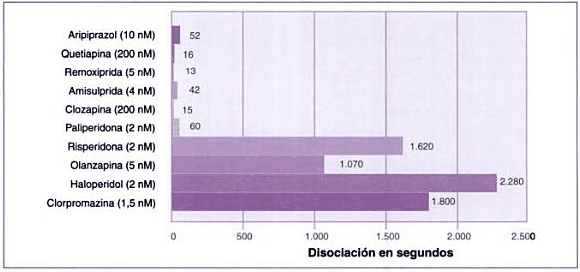
Velocidad de disociación de diversos antipsicóticos del receptor D2 (50%). Modificada de Seeman (2005).
Fuente del gráfico: Libro "Las esquizofrenias: sus hechos y valores clínicos y terapéuticos" 2007 de Alfonso Chinchilla Moreno.
Imagen:
Todos los fármacos antipsicóticos disponibles actualmente se unen a los receptores de dopamina. Los antipsicóticos típicos se unen fuertemente y disocian lentamente del receptor de la dopamina, mientras que los fármacos atípicos se unen más libremente y disocian más rápidamente.
|
D1 |
D5 |
D2 |
D3 |
D4 |
Agonistas |
skf 38393 |
Dopamina |
Bromocriptina |
Dopamina |
Dopamina |
Antagonistas |
Alfa-Flupentixol |
|
Haloperidol |
Sulpiride |
Clozapina |
Distribución |
N. Estriado |
Hipotálamo |
N. Estriado |
T.olfatorio |
CORTEX FRONTAL |
Ejemplo de receptores unidos a proteínas G estimulando a la Adenilato Ciclasa (Gs)
Hay otro tipo de transducción con proteínas G que estimula a la fosfolipasa C (PLC) (G0), con la aparición de IP3 y DAG como segundos mensajeros: verlo en estimulantes apartado varios.
Antagonista D2
Los receptores D1 son responsables de mantener a los receptores D2 en un estado de desensibilización. Este control parece estar perdido en los trastornos psicóticos. En esquizofrenia no se han llevado a cabo estudios para evaluar el potencial efecto antipsicótico de agonistas D1.Actualmente se aceptan dos receptores dopaminérgicos centrales: D
1 y D
2, y otros dos receptores dopaminérgicos periféricos: DA
1 y D
A2. Los receptores D
1 son postsinápticos y los D
2 postsinápticos y presinápticos. Los receptores D
1 utilizan el segundo mensajero (AMPc) para su actuación y su consecuencia immediata es la activacion de la adenilatociclasa. Sin embargo, los receptores D
2 inhiben la actividad de la adenilatociclasa. Es, por tanto, indudable que las consecuencias de su alteración funcional, así como de la actividad de agonistas y antagonistas dependerá de la densidad de receptores de uno u otro tipo que se presenten en las estructuras encefálicas y del umbral de sensibilidad que presentan en respuesta a la dopamina. Parece ser que los receptores D
1 son mayoritarios en las proyecciones nigroestriadas y mesolímbicas y que las D
2 lo son en las proyecciones mesocorticales. Los receptores D
1 modifican sus umbrales y presentan una mayor sensibilidad en la esquizofrenia y no en el Parkinson; luego sus antagonistas podrían tener un buen efecto antipsicótico sin alterar la regulacion motora. La densidad de los receptores D
2 es mayor en la esquizofrenia y en el Parkinson, por lo que sus antagonistas podrían presentar fuertes efectos antipsicóticos, pero también extrapiramidales. A nivel periférico, el receptor DA
1 es postsináptico y el receptor DA
2 es presináptico o autorreceptor.
Antagonista D1
Fuente: http://www.cnsforum.com/imagebank/section/Dopaminergic/default.aspx
http://www.cnsforum.com/educationalresources/imagebank/
El receptor beta adrenérgico se comunica con el citoplasma estimulando una segunda proteína que se conoce como proteína G por razones que enseguida se entenderán. La proteína G generalmente se encuentra cerca del receptor en forma inactiva. Cuando el receptor es activado por un ligando , rápidamente actúa sobre la proteína G.
La proteína G responde rápidamente activándose a si misma y, ya en el estado activo, envía señales a otras moléculas del citoplasma celular. Sin embargo la proteína G solo permanecerá en estado activo por un breve periodo pasándose luego a la forma inactiva.
En efecto, la proteína G actúa como una llave binaria autoapagable ( equivalente a una llave de luz autoapagable como las que se encuentran en muchos edificios ), que cuando esta "on" permanece así un pequeño periodo de tiempo hasta que se pone asimismo en "off".
Los dos estados (encendido/apagado) están determinados por el nucleótido de guanina que se encuentra pegado a ella (de allí el nombre de proteína G).
Cuando esta inactiva pega GDP (guanosin difosfato), cuando esta activa GTP (guanosin trifosfato) En consecuencia, la forma inactiva ("apagada") de la proteína G se presenta cuando la misma tiene ligado GDP.
Cuando el ligando se "pega" a ella, libera GDP y permite que el GTP se una. Esta forma de la proteína G que liga GTP, es la forma activada ("encendida") de la misma Cuando esta "encendida" libera señales al interior de la célula. Luego de un corto periodo (segundos o menos) la proteína G hidroliza el GTP a GDP y se "apaga" asimismo.
Esta hidrólisis representa un mecanismo de retroalimentación negativo que asegura que la proteína G solo estará activa por un corto tiempo.
EFECTO CASCADA
Se describirá brevemente el flujo de la señal ( a menudo nombrado como efecto cascada) que es disparado por la proteína G activa. La proteína G esta formada por tres subunidades proteicas llamadas alfa, beta y gamma. En su forma inactiva las tres subunidades se encuentran unidas. La subunidad alfa es la que tiene el GDP. Cuando el receptor beta adrenérgico activa la proteína G, la subunidad alfa libera el GDP, pega GTP y luego se separa de las subunidades beta y gamma.
Cuando esto ocurre la subunidad alfa pierde su afinidad por el receptor, se disocia de el, y se mueve hacia otra proteína cercana, la enzima adenilato ciclasa, que hasta el momento estaba inactiva y que ahora es activada y comienza su trabajo: convertir el ATP en 3'5' AMP cíclico. Esta reacción implica liberar los fosfatos gamma y beta del ATP ligar el fosfato restante (que esta esterificando a la ribosa en la posición 5') al hidroxilo 3' formando una estructura cíclica conocida como "AMP cíclico" o simplemente AMPc.
Luego de varios segundos de la unión con la adenil-ciclasa, la subunidad alfa de la proteína G hidroliza el GTP , abandona la adenilato ciclasa y se pone en "off" (apagado) y retorna a su unión con las subunidades beta y gamma (lugar de donde había "desertado" al comienzo del "juego"). La adenil ciclasa se torna inactiva y deja de producir AMPc.
Todo este ciclo origina un breve "pulso" de señales que producen, en este caso, unos cientos de moléculas de AMPc.
El AMPc actúa como un segundo mensajero que difunde por el citoplasma (el primer mensajero es el ligando en la superficie celular, estos ligandos son en general productos conocidos como hormonas: por ejemplo la epinefrina) llevando su acción al mismo. En ausencia de eventos como el señalado anteriormente, el nivel de AMPc en muy bajo, se eleva como consecuencia de este proceso y, como puede Ud. imaginarse, es rápidamente hidrolizado a AMP (no cíclico) con lo cual el estimulo termina.
En varios sitios de la célula el AMPc se pega a otra enzima una serina/treonina proteina-kinasa, llamada la AMPc-kinasa dependiente o simplemente ''A kinasa''. La A kinasa, una vez activada , puede fosforilar específicamente determinadas proteínas , específicamente en sus residuos serina/treonina, y al hacerlo las activa. En el caso de las células del hígado que almacenan grandes cantidades de glucógeno , el AMPc hace que la A-kinasa se active y
- fosforile y por lo tanto active una enzima que activa la glucogeno-fosforilasa, que a su turno rompe el glucógeno en moléculas de glucosa-1-fosfato; y
- fosforila la glucógeno sintetasa, y de esta manera haciendo que la misma no trabaje, e impidiendo la reconversión del glucosa en glucógeno. Estos dos cambios (activación de la enzima que degrada, e inhibición de la enzima que sintetiza) aseguran la movilización del glucógeno almacenado en el hígado a glucosa.
Otras reacciones son activadas de manera similar en otras células para contribuir a la respuesta de alerta/huida del organismo.
Esta "cascada " de reacciones aumenta enormemente la señal que llego a la célula.
Una molécula de epinefrina causa la activación de la subunidad alfa del receptor, ella activara a una adenilato-ciclasa, la cual a su vez sintetizara cientos de AMPc. Cada uno de los AMPc puede activar una kinasa- AMPc - dependiente, que a su vez puede modificar cientos de moléculas en la célula..
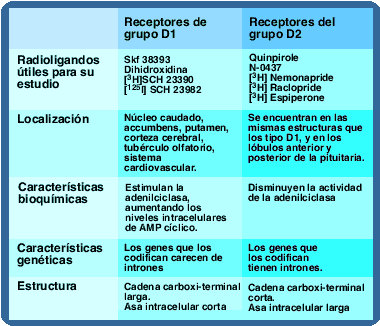
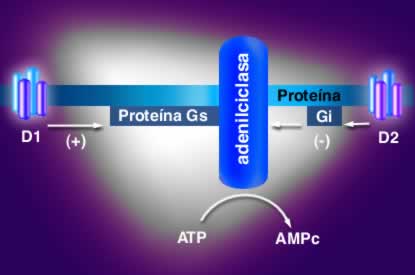
Acción de los receptores tipo D1 y D2 sobre la adenilciclasa.
Todos los receptores dopaminérgicos son ligados a proteína G, la diferencia entre los dos grupos radica en el tipo de proteína G y los agentes mensajeros utilizados. Todos los receptores ligados a proteína G constan de una porción extracelular que tiene el grupo amino-terminal, una porción proteínica que atraviesa la membrana 7 veces y una porción intracelular que posee la terminación carboxilo que con frecuencia e asocia a un grupo palmitol.
Existen diferencias en la longitud de las asas intracelulares y las terminaciones carboxílicas entre los receptores tipo D1 y los receptores tipo D2, en los primeros las asas intracelulares son cortas, y las terminaciones carboxílicas largas, mientras los D2 tienen asas intracelulares largas y terminaciones carboxílicas cortas. Adicionalmente, existen diferencias entre los genes que los codifican los de receptores D1 carecen de intrones. También estimulan la síntesis de AMP cíclico mediada por proteínas asociadas a nucleótidos de guanina llamadas proteínas G, del subtipo estimulatorio (Gs).
Ambos tipos de receptor son muy comunes en el núcleo caudado, putamen y núcleo accumbens. Los receptores D1 se encuentran además en neuronas postsinápticas del estriado, la región medial del globo pálido y en la parte reticulada de la sustancia nigra.
La mayoría de receptores D2 en el estriado actúan como autorreceptores. Estudios post mortem de pacientes esquizofrénicos han mostrado aumento en la densidad de receptores D2 a este nivel, lo que probablemente se debe al tratamiento crónico con antipsicóticos pues los estudios imagenológicos in vivo de pacientes que no han recibido tratamiento farmacológico nunca, no fueron consistentes con este hallazgo.
En el hipocampo, los receptores D1 inducen la producción tardía de proteínas, responsable de los mecanismos de potenciación del estímulo.
Los receptores D2 están asociados a proteínas G del subtipo inhibitorio (Gi).
La activación de los receptores tipo D1 produce activación de adenilciclasa, aumentando las concentraciones de AMP cíclico , responsable del aumento marcado de la concentración de calcio intracelular, que al parecer depende de liberación del elemento de las reservas intracelulares. Este fenómeno es importante en la liberación de dopamina al espacio sináptico.
Por otra parte, la activación de los receptores tipo D2, inhibe adenilciclasa, estimula a mitogénesis y produce acidificación del medio extracelular. Estos receptores D2 son más comunes en el cerebro humano que los D1.
La mayoría de los fármacos antipsicóticos tiene mayor afinidad por uno de los 2 grupos. Las tioxantinas y fenotiazinas tienen mayor afinidad por los receptores D1, mientras las benzamidas y butirofenonas prefieren los receptores D2. La clozapina muestra una moderada selectividad sobre los receptores denominados antiguamente D4, que pertenecen al grupo tipo D2.
Enlaces:
Hay un dibujo en la sección varios 2 apartado COREAS que de un vistazo se capta la explicación de cómo funciona el modelo de los antipsicóticos bloqueo D2 del Globo pálido externo de la vía indirecta
Funcionamiento de los núcleos de los ganglios de la base
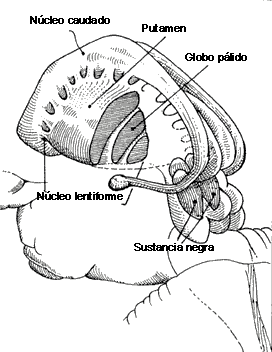
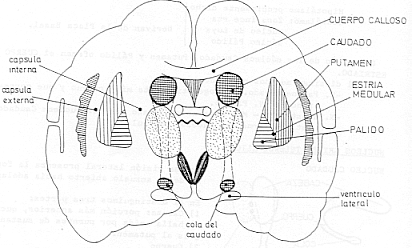
Es muy difícil estudiar algo que se origina en la corteza cerebral y que acaba volviendo a ella, pero luego además hay superpuestos otros lazos. Una de las cosas importantes es que hay una vía directa de acción sobre la corteza o en el caso de la sustancia negra sobre el tubérculo cuadrigémino superior. La corteza sobre todo las áreas de asociación frontales y parietales activan al estriado, putamen y caudado; el estriado inhibe al pálido y el segmento interno del pálido inhibe a su vez al tálamo, el cual excita a la corteza motora.
Esta es la vía directa por la cual la corteza vuelve a actuar sobre la corteza. La entrada es fundamentalmente de la corteza de asociación aunque no únicamente ya que la corteza motora también proyecta a caudal y, la salida es fundamentalmente a corteza motora; principalmente a la corteza premotora porque estamos hablando de la iniciación de los movimientos sobre todo, y es ésta quien planifica los movimientos. La causa del Parkinson es la entrada dopaminergica que surge de la parte compacta de la sustancia negra y que va al caudado, y que sobre esta vía esta excitando. El Parkinson está producido por la lesión de las vías dopaminergicas, pero es difícil explicar eso porque la dopamina tiene que inhibir y excita al mismo tiempo. Esto ocurre ya que las misma neuronas dopaminergicas que proyectan al caudado actúan sobre distintas células espinosas que tienen receptores a dopamina diferentes. Entonces estas las que proyectan a la vía directa al pálido interno tiene receptores a dopamina de tipo D1 y estos receptores provocan la excitación de esas células, despolarizando esas células cuando se libera dopamina por esas sinapsis. En cambio sobre este circuito están añadidos los circuitos indirectos. La vida indirecta con entradas al caudado, putamen, a otras células estriadas espinosas pero no las mismas de la vida directa. Éstas proyectan al segmento externo del pálido. La proyección de esas células dopaminérgicas tiene una acción inhibitoria sobre sus receptoras, ya que tienen receptores D2 y cuando la dopamina se fija en esas células se hiperpolarizan. La misma dopamina proveniente de la sustancia negra que excita la vida directa, inhibe la vía indirecta.
El Parkinson se explica mejor ya que se sabe que la dopamina excita e inhibe diferentes vías, La proyección indirecta desde el estriado al pálido externo es también inhibitoria Ahora el pálido externo proyecta por axones gabaergicos inhibitorios al pálido interno y al núcleo subtalámico El núcleo subtalamico su salida es activar el pálido externo.
La activación de la vida directa provoca la liberación de la corteza de la inhibición consiguiendo así la activación de la misma. En la vida indirecta se activa la corteza de asociación, se activa el caudado-putamen, éste inhibe al pálido externo por lo que actúa menos, inhibiendo menos el pálido interno, el cual en estas condiciones actúa inhibiendo el tálamo que inhibe la corteza. El núcleo subtalámico es el mismo efecto, cuando activa la vía indirecta se opone a la acción de la vida directa, la via indirecta inhibe la corteza cuando se activa, Tenemos un sistema de freno y aceleración.
Uno facilita la activación de la corteza motora, y el otro esta impidiendo que se active la corteza motora. Y los desbalances de esta función provocan que la patología de los ganglios de la base en unos casos predomine la acción de la liberación de la corteza con movimientos involuntarios; en el otro caso predomine la acción del freno de la corteza con la torpeza de movimientos característicos del Parkinson.
(Texto resumen muy básico y esquemático que para entenderlo hay que leer con detenimiento los enlaces de abajo)
VÍA DOPAMINÉRGICA
Las neuronas de la parte compacta de la sustancia negra son origen de una vía dopaminérgica que actúa sobre el caudado y putamen.
Las neuronas del caudado y putamen que proyectan al globo pálido interno y parte reticular de la sustancia negra tienen receptores para la dopamina de tipo D1, que son activadores, por lo que la dopamina activa la vía directa estimuladora de los movimientos.
Las neuronas del caudado y putamen que proyectan al globo pálido externo tiene receptores para la dopamina de tipo D2, que son inhibidores, por lo que la dopamina inhibe a la vía indirecta inhibidora del movimiento.
La dopamina por tanto, estimula el movimiento por las dos vías, porque estimula la vía estimuladora e inhibe a la vía inhibidora.
Enlaces:
Sustrato neural
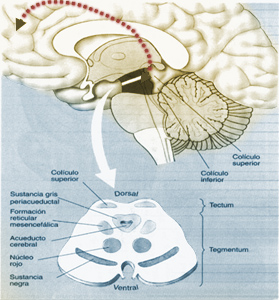
Los ganglios de la base (GB) participan en el procesamiento de señales motoras cognitivas y afectivas, motivo por el cual, la estereotipia puede incluir dichos fenómenos (motores/cognitivos) en su expresión clínica(3).
La estimulación dopaminérgica puede lograrse por el uso de psicoestimulantes que sin ser agonistas directos del receptor dopaminérgico, logran aumentar la biodisponibilidad de dopamina (mecanismo indirecto).
La acción se produce en el espacio sináptico a través de un aumento de la liberación presináptica (anfetamina) o por la inhibición de la recaptación presináptica de dopamina (cocaína).
Por otro lado, la estimulación dopaminérgica puede lograrse a nivel post-sináptico (mecanismo directo), estimulando al receptor dopaminérgico con agonistas de la familia de receptores D1 y D2 (apomorfina).
El cuerpo estriado es parte integrante de los ganglios de la base (Fig 1). Recibe aferencias desde la corteza cerebral a través de las fibras glutamatérgicas cortico-estriatales, que hacen sinapsis en la cabeza de las espinas dendríticas de las neuronas gabaérgicas medianas espinosas de proyección (MSSN)(2). Éstas conforman el 90% de la población celular del cuerpo estriado(21), y formalizan las eferencias del estriado al proyectar sobre otras estructuras de ganglios basales que luego se dirigen al tálamo. El estriado tiene dos salidas o eferencias, que se conocen como camino directo a la sustancia nigra pars reticulada (SNr) - globo pálido interno (GPi) y el camino indirecto a dichos núcleos(13). Si bien el receptor dopaminérgico D1 se encuentra en mayor densidad en el camino directo y el receptor D2 en el camino indirecto, ambos pueden ser hallados en los dos grupos neuronales gabaérgicos de proyección.
Dichos receptores se alojan en el cuello de las espinas dendríticas de la neurona gabaérgica, neuromodulando tanto a la transmisión gabaérgica como también a la glutamatérgica que sobre ellas hace sinapsis desde las aferencias cortico-estriatales al nivel de las cabezas de las espinas dendríticas de las MSSN(15). En el camino directo, el receptor D1 activa la adenilatociclasa, promoviendo un mayor tono gabaérgico que finalmente promoverá el movimiento, a través de un patrón fásico. En el camino indirecto el receptor D2 que inhibe a la adenilatociclasa, inhibe a dicha vía. La vía indirecta tiene como función la de inhibir el movimiento a través de un patrón tónico.
Por lo tanto la dopamina ejerce una acción deshinibitoria en el estriado que promueve el pasaje de señales desde el tálamo a la corteza cerebral(14).
Durante el neurodesarrollo(53) las neuronas estriosomales son las primeras en nacer. Son así precursoras del estriado y con su presencia determinarán el sustrato neural gabaérgico del estriado con mayor conexión dopaminérgica proveniente de las proyecciones más ventrales de la sustancia nigra pars compacta (SNc). A su vez las eferencias gabaérgicas se dirigen a la SNc constituyendo un freno a la neurotransmisión dopaminérgica (feed-back negativo estriato-nigral).
***
Circuito Límbico
Desde la corteza orbitofrontal medial (COFm) y la corteza cingulada anterior (CCA)(55), se proyecta al estriado ventral: caudado ventral, núcleo accumbens (na) y tubérculo olfatorio (to).
Por estudios realizados con PET conjuntamente a evaluaciones neuropsicológicas, se consideran en la corteza cingulada anterior (CCA) dos regiones bien delimitadas funcionalmente. La anterior ligada a procesamiento afectivo y ejecutivo, y la posterior, vinculada a aspectos cognitivos(10,11,18). La corteza orbitofrontal (COF) se activa en el procesamiento afectivo y motivacional. La proyección glutamatérgica de ambas (CCA y COF) es hacia el estriado ventral cuya principal estructura es el núcleo accumbens y el caudado ventral(52).
La CCA y la COF establecen sinapsis con el compartimento estriosomal del caudado ventral, que determina el link entre las distintas señales motoras y cognitivas. A su vez, el caudado ventral recibe aferencias dopaminérgicas provenientes del área tegmental ventral (ATV), cuya activación, ligada a los fenómenos de gratificación, la constituye en una vía sumamente importante para estudiar fenómenos de plasticidad neuronal en las adicciones, y en la sintomatología positiva y negativa de la esquizofrenia. El estriado ventral (núcleo caudado, núcleo accumbens y tubérculo olfatorio) proyecta al pálido ventral (PV). Luego el PV se conecta con el tálamo y posteriormente por las fibras tálamo-corticales se cierra el loop, volviendo nuevamente hacia la corteza(25,52).
Las alteraciones de este circuito están asociadas a anhedonia, inhibición psicomotriz, mutismo(3,62).
Circuito prefrontal dorsolateral (CPF-DL)
Es un circuito vinculado al procesamiento del componente espacial y a la constitución de la working memory, que se realiza fundamentalmente por la corteza pre-frontal dorso-lateral (CPF-DL)(31) y la corteza parietal posterior (CPP) en una interacción cortico-cortical(47,49,50,59). A su vez la CPF-DL envía sus proyecciones glutamatérgicas a la región dorsolateral del caudado participando en las funciones ejecutivas y de planificación del movimiento junto al área suplementaria motora (ASM)(3).
Recordaremos que las dos neurotransmisiones rápidas del SNC que operan a través de canales iónicos son la glutamatérgica y la gabaérgica que se modulan entre sí(64). A su vez reciben fibras provenientes de regiones subcorticales que poveen distintos neurotransmisores modulatorios de ambas(6). Entre ellos se encuentra la serotonina, la dopamina, la acetilcolina y la noradrenalina(14,15,27). Toda variación del tono de cada uno de los transmisores mencionados, influirá en el patrón de disparo de la neurona glutamatérgica(1,16). En la enfermedad de Parkinson, donde se produce un menor tono dopaminérgico, se desencadena un mecanismo de plasticidad neuronal por el cual hay una up regulation del receptor D2 por déficit en la biodisponibilidad del agonista en el espacio sináptico. Cuando la disminución del tono dopaminérgico se produce por el bloqueo del receptor D2, también se produce la up regulation del receptor D2 que se acompaña de una down regulation del receptor D1 que también se expresa en la CPF además de la expresión estriatal(45). De esta forma, dichos cambios se acompañan de perseveraciones o estereotipias cognitivas y motoras.
La terapéutica que se implementa en la esquizofrenia, consistente en lograr un bloqueo dopaminérgico en el estriado, no es la única gatilladora de la sintomatología negativa. En todo caso se puede sumar al compromiso cognitivo vinculado al déficit en la malla de conectividad sináptica a nivel de la CPF(6,8,31). Se ha estudiado su conformación histopatológica, reportándose una disminución del neuropilo. Este déficit que no significa pérdida neuronal ni gliosis reactiva, consiste en una menor cantidad de contactos sinápticos en la corteza, y podría ser la razón de la sintomatología negativa que se verifica en la esquizofrenia. Por lo tanto, la CPF-DL que se activa en momentos de la evaluación espacial y es la principal estructura activada en los momentos en que se produce la working memory o block de apuntes, procesa en serie con la CPP en la dirección cortico-cortical, así como también con el estriado dorsal (principalmente el núcleo caudado en su porción dorsolateral) en la dirección cortico-subcortical, generando una integración en paralelo del procesamiento cognitivo, espacial y motor(23,34,35,37,39).
***
Importancia del estriado dorsal y ventral como ductus entre el aspecto motor y el afectivo. Procesamiento en paralelo
Si bien el circuito motor y el límbico, parten de distintas localizaciones corticales, dado que el motor lo hace de áreas motoras y el límbico desde la corteza cingulada y corteza orbitofrontal medial, dichas estructuras corticales proyectan al estriado. La motora sobre el estriado dorsal (fundamentalmente el putamen) y la límbica sobre el estriado ventral (fundamentalmente el caudado ventral y núcleo accumbens). Si bien ambos circuitos parten de distintas cortezas, al pasar por el estriado primero, y por el globo pálido luego, convergen y comparten estas últimas estructuras para llegar al tálamo. Justamente desde el tálamo les vuelve a las cortezas el procesamiento de la señalización, constituyendo el "spot" de luz en los modelos teatrales(3,4,7). De esta manera, el movimiento que se lleva a cabo por el sistema piramidal es supervisado, motivado, rectificado o ratificado por el sistema extrapiramidal(41,42,63).
Ello significa que por dicha estructura pasan distintas señales que serán decodificadas como motoras, afectivas y motivacionales respectivamente. Estos circuitos son interdependientes, procesan distintos aspectos del movimiento: la programación, la motivación de realizarlo, la ejecución y el requerimiento cognitivo necesario(3,41,42).
***
Cuando la acción farmacológica consiste en la disminución del tono dopaminérgico como ocurre con el uso al largo plazo de antagonistas dopaminérgicos (haloperidol)(5) así como en la enfermedad de Parkinson (neurodegeneración nigro-estriatal), se produce incremento en la expresión genómica de c-fos a nivel estriatal con mayor disponibilidad del neuropéptido encefalina, sobre el camino indirecto, donde la densidad del receptor dopaminérgico D2 es mayor(5,26). Si recordamos que la acción del receptor D2 es la de inhibir a la enzima adenilatociclasa, el bloqueo de dicho receptor por antipsicóticos típicos, genera una contra-acción. De esta manera se activan las neuronas gabaérgicas de proyección de la vía indirecta cuyo co-transmisor es el neuropéptido encefalina (MSSN-encefalina). Por estimulación glutamatérgica cortico-estriatal, la vía indirecta mantiene a través de un patrón tónico, una inhibición sobre el tálamo y por lo tanto limita el pasaje de información irrelevante ("ruido"), permitiendo la jerarquización de las señales cognitivas y motoras hacia la vía tálamo-cortical. La dopamina proveniente de la vía nigro-estriatal, a través del receptor D2 inhibe a asta vía(13). Cuando se produce un aumento desmedido del tono dopaminérgico (modelo de la psicosis anfetamínica)(24), se permite el pasaje de «ruido» a la corteza, por insuficiencia del filtro talámico para seleccionar y constituir señales(14,15). El haloperidol activa dicha vía por bloqueo del receptor D2, incrementando la cadena de eventos intracelulares, vía AMPc con el consecuente aumento de c-fos estriatal(25,26).
La producción de c-fos está ligada a actividad celular. Por lo tanto, el bloqueo del receptor dopaminérgico D2, activa al camino indirecto inhibiendo el pasaje de información irrelevante a la corteza cerebral. Si bien así se mejora el trastorno del pensamiento por neutralizar la "ensalada de palabras" a través de la limitación del pasaje de información no relevante, también frena el pasaje de señales motoras, limitando el movimiento con la finalidad de neutralizar la "excitación psicomotriz". En el momento de un exceso de dicha inhibición, se produce el síndrome deficitario por neurolépticos, que provoca lentitud del movimiento ("bradikinesia") y lentitud de la velocidad del pensamiento ("bradipsiquia") con perseveraciones.
La bradikinesia puede cursar con temblor y perseveraciones motoras, que en el uso crónico y sobre todo cuando fueron utilizados los anticolinérgicos centrales en el largo plazo (trihexifenidilo/biperideno), la diskinesia tardía puede ser el cuadro residual persistente. Si bien falta mayor confirmación sobre la posible localización del receptor dopaminérgico D2 sobre la terminal glutamatérgica cortico-estriatal, es probable que su función, al ser activado por la dopamina proveniente de la vía nigro-estriatal, consista en limitar la liberación de glutamato sobre la neurona mediana espinosa gabaérgica (MSSN). El bloqueo persistente del receptor dopaminérgico D2, generaría un aumento persistente en la liberación de glutamato, por modificación en los patrones de disparo neuronal(66). El glutamato es un aminoácido excitatorio vinculado a eventos de neuroplasticidad óptima en el SNC, siendo un neurotransmisor necesario para los fenómenos de aprendizaje y memoria(48). Sin embargo, también está vinculado a eventos de neurotoxicidad a través de un aumento en la conductancia al Ca++(5,66).
En el estriado, el bloqueo crónico del receptor dopaminérgico D2 también se acompaña de una up-regulation del receptor adenosínico A2a, que provoca una contra-acción del receptor dopaminérgico D2 y un desacople de la dopamina al receptor D2. Además de una up-regulation del receptor D2 a nivel estriatal, se produce una down-regulation del receptor D1, incluso a nivel de la corteza prefrontal, generando por dicho mecanismo un déficit cognitivo simultáneo al trastorno motor(45). En el estriado el receptor dopaminérgico D1 se encuentra en las espinas dendríticas de las neuronas gabaérgicas medianas espinosas cuyo co-transmisor es la dinorfina (MSSN-din/sustancia P). Este es el camino directo a la SNr/GPi, generador de movimiento dado que el receptor D1 activa a la adenilatociclasa y por dicho mecanismo intracelular activa el camino directo. Cuando la down regulation del receptor D1 es a nivel estriatal, disminuye el movimiento por menor tono del camino directo.
A su vez el receptor dopaminérgico D1 se encuentra en las terminales estriato-nigrales de las neuronas gabaérgicas de proyección, al parecer aumentando la liberación de GABA en la SNr(61). La down-regulation del receptor D1 antes mencionada, puede ser causal de un menor tono gabaérgico sobre el SNr, el cual desencadena como respuesta una up-regulation de receptores al GABA en SNr. El menor tono gabaérgico sobre SNr, deja a éste último con mayor tono inhibitorio gabaérgico en su proyección sobre el tálamo, alterándose el patrón de disparo y generándose un disbalance en todas las neuromodulaciones de GB(22).
Conclusiones
Importancia de la conexión nigro-estriosomal-corteza cingulada.
Con el uso crónico de psicoestimulantes(36) o con el tratamiento crónico con L-Dopa en la enfermedad de Parkinson en forma intermitente(32), se puede producir plasticidad neuronal, con cambios en la expresión genómica, sobre todo en la vía directa de salida del estriado. El aumento en la producción de IEG (inmediate-early genes) para la codificación de los factores de transcripción de la familia Fos-Fra no se verifica en igual proporción en los dos compartimentos del estriado, compuesto por los estriosomas y la matriz.
La activación genómica es generalmente mayor en el compartimento estriosomal. El cambio en dicho compartimento se realiza a expensas de un menor cambio en el matriosomal. La mayor plasticidad verificada en los estriosomas por sobre la matriz, es predictor del desarrollo de estereotipias(12). Esto se debe a la conexión con la sustancia nigra pars compacta (SNc) y que constituye el camino nigro-estriosomal. La dopamina proveniente de la SNc influirá sobre las neuronas gabaérgicas estriosomales. A su vez las neuronas estriosomales constituyen el link con el sistema límbico, jerarquizándose la participación de la corteza cingulada anterior(33). Es así como al estar vinculada con la gratificación o reward la dopamina genera memorias que promoverán la repetición(60).
La estereotipia no constituye un movimiento completo o acción específica. Son repeticiones de fragmentos de movimientos específicos. Es necesario recordar que el compartimento matriosomal (M) del estriado, está conectado con la corteza motora, corteza sensorial y corteza asociativa y es el circuito más importante para la planificación, programación, secuenciación y ejecución motriz. Justamente el compromiso matriosomal por los fenómenos de sensibilización estriosomal, parece ser decisivo para la generación de estereotipias. A su vez, se recordará que la conexión cortical del compartimento estriosomal es con la corteza cingulada anterior, la corteza orbitofrontal y la corteza prefrontal medial.
En el caso del bloqueo dopaminérgico con antipsicóticos típicos en el largo plazo, se pueden generar cambios genómicos en el camino indirecto y ser causales de diskinesia tardía, donde la estereotipia motora puede persistir a lo largo de la vida.
Por lo tanto, por los cambios moleculares descriptos para la producción de estereotipias, se establece un disbalance entre las conexiones cortico-estriato-tálamo-corticales y los distintos caminos de salida estriatal(51), tanto para el directo como para el indirecto, reflejados por el uso de agonistas como por antagonistas dopaminérgicos respectivamente.
Dicho disbalance, permite verificar que en el SNC no se producen reacciones lineales, (19) dada la multiplicación de efectos en cascada que se producen por las diferentes neuromodulaciones y la neuroplasticidad.
Para leerlo entero:
http://www.alcmeon.com.ar/9/36/Fadel.htm
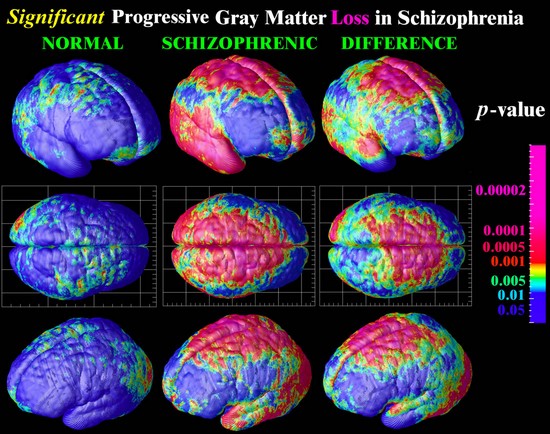
La pérdida de tejido cerebral en un intervalo de cinco años, la pérdida grave se muestra en color rosa. Los pacientes con esquizofrenia (derecha), más rápido pierden tejido cerebral que sus compañeros sanos (izquierda).
Imagen cortesía de Paul Thompson, Laboratorio de Neuro Imaging, la Universidad de California.
http://www.isgtw.org/?pid=1000520
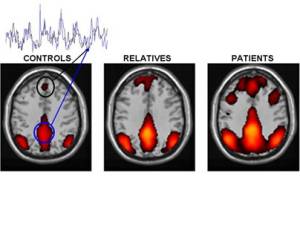
Alteración de la conectividad del cerebro en personas con esquizofrenia y familiares de primer grado. Zonas de color representan una red interconectada de regiones del cerebro que muestran una actividad sincronizada (superposición de huellas en color negro y azul) cuando los sujetos descansan y permiten que su mente vague. El importe de la sincronía, que refleja la fuerza de las conexiones funcionales entre los distintos ámbitos, está aumentada en pacientes con esquizofrenia. Los parientes en primer grado de las personas con la enfermedad también muestran un cierto aumento, aunque menos que los pacientes. Círculo Negro: la corteza prefrontal medial. Círculo Azul corteza cingulada posterior / precuneous. Crédito de la imagen: Susan Whitfield-Gabrieli, McGovern Institute for Brain Research del MIT
http://www.schizophreniaforum.org/new/detail.asp?id=1486
Los voluntarios fueron escaneados mediante resonancia magnética funcional por imágenes (fMRI), mientras descansaban y mientras ejecutaban tareas de memorización fáciles o difíciles. El punto de vista tradicional sobre la esquizofrenia es que los pensamientos, percepciones y emociones alteradas que caracterizan la enfermedad están causados por desconexiones entre las regiones cerebrales que controlan estas diferentes funciones.
Pero este estudio ha desvelado que la esquizofrenia también involucra un exceso de conectividad entre las regiones cerebrales involucradas en la reflexión sobre uno mismo y que devienen activas cuando no estamos pensando sobre nada en particular, o cuando pensamos sobre nosotros mismos.
Normalmente, las personas suprimen este sistema cuando realizan tareas que demandan mucha atención, pero Susan Whitfield-Gabrieli (MIT), John D. Gabrieli (MIT), Larry J. Seidman (Escuela Médica de la Universidad de Harvard), y los otros 10 autores del estudio, encontraron que los pacientes con esquizofrenia no actúan de esta manera.
http://www.amazings.com/ciencia/noticias/040309c.html
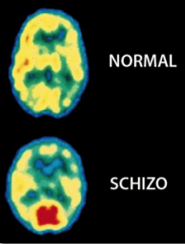
Tomografía por Emisión de Positrones (TEP) del cerebro muestran grandes diferencias en la distribución de la actividad eléctrica en un cerebro normal, comparado con el cerebro de un paciente esquizofrénico (derecha). La esquizofrenia afecta a aproximadamente 1% de la población mundial. Reproducido con permiso de. Photo Researchers, Inc.
Todas las diferencias Neuroanatómicas y funcionales de la Esquizofrenia
http://www.schizophrenia.com/family/disease.htm
ESQUIZOFRENIA ENLACES:
- Psicopatologías. Dismetria cognitiva. Trastornos hipocampales. Fosfolipasa. Cerebro farmacología. Estados psicóticos. Neuropsiquiatría
- farmacología. Síntomas. Tipos de Esquizofrenia. Causas. Daños Cerebrales. Fármacos Antipsicóticos
- Dopamina y conducta
- ANTIPSICÓTICOS DE LA UNIVERSIDAD AUTÓNOMA DE BARCELONA
- http://cuentospintados.com/portfolio/geodon/centro_enf/art1a.htm
- PERSPECTIVA NEUROBIOLÓGICA DE LA ESQUIZOFRENIA
- Aspectos neurobiológicos de la esquizofrenia
- http://www.schizophreniaforum.org/
- http://www.schizophrenia.com/
- http://yaletomorrow.yale.edu/news/schizophreniaresearch.html
Afinidad al receptor D2 de los antipsicóticos:
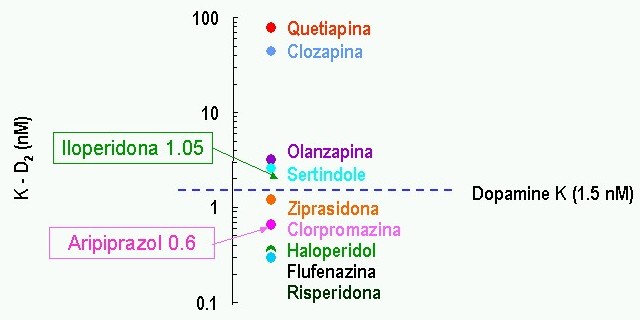
Lawler C, et al. Neuropsychopharmacology 1999; 20:612-27; Corbett R, et al. CNS Drug Reviews 1997; 3:120-47 El mismo gráfico pero más completo
(adaptado de Zito 1994 & Kane 1996)
Gráficos sacados de psicofarmacologia.info

Gráficos de http://www.msc.es/estadEstudios/publicaciones/docs/20001-01.pdf
Perfil Antipsicótico
Nombre |
Potencia antipsicótica |
Sedación |
Efectos vegetativos |
Síntomas extrapiramidales |
|
|
|
|
|
Leyenda: (+) muy leve, + leve, +(+) moderado, ++ intenso, ++(+) potente, +++ muy potente
* Fuente de la búsqueda: farmacología humana, Jesús Flórez
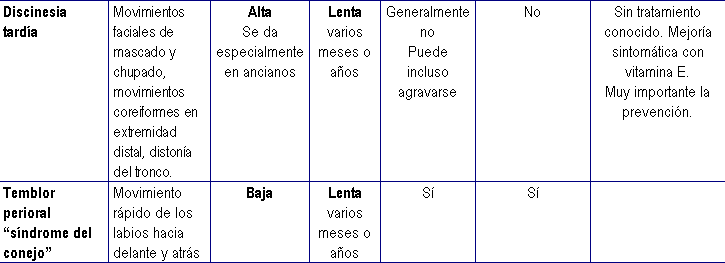
Principales reacciones extrapiramidales por antipsicóticos
Fuente:http://www.bipolarweb.com/ANTISICOTICOS.htm
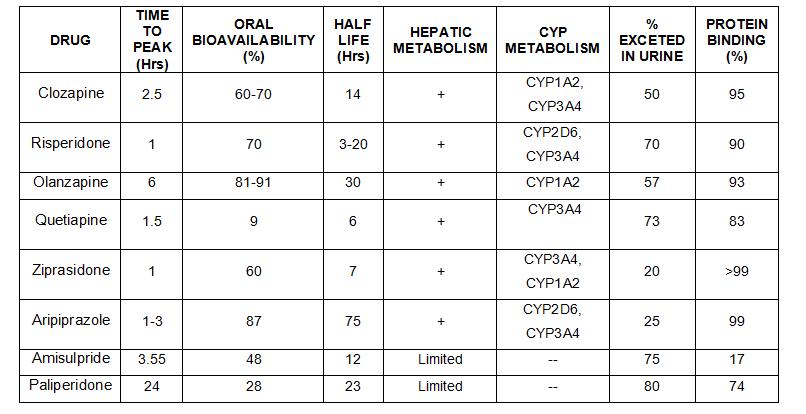

FARMACOCINETICA
CARACTERISTICAS FARMACOCINETICAS DE LOS ANTIPSICOTICOS
- Absorción
- Vía oral: pico máximo plasmático, 2-4 horas (absorción modificada por café, té, anticolinérgicos, antiácidos).
- Vía intramuscular: pico máximo, 20-30 min.
- Se desaconseja la vía intravenosa.
- Distribución
- Gran variabilidad interindividual de niveles plasmáticos (entre 10-100 veces; efecto del primer paso hepático).
- Unión a proteínas plasmáticas en un 90-98%.
- Alta lipofilia. Alta biodisponibilidad. Afinidad preferente en SNC, pulmones, tejidos altamente vascularizados.
- Eliminación
- Metabolización preferente en microsomas hepáticos:
- Glucuronoconjugación
- Hidroxilación
- Sulfóxidos
- Eliminación renal:
- 1/2 a (Vida media de distribución): 2 horas (oral)
- 1/2 ß (Vida media de eliminación): mayor de 30 horas (oral)
Su absorción tras la administración oral es rápida (2-4 horas) pero susceptible a alteraciones por diferentes factores como son los antiácidos, los anticolinérgicos, el café y el té, que actúan retardando su absorción. Se recomienda el intervalo de 2 a 4 horas entre el consumo de estos productos y la administración de un antipsicótico. La absorción por vía intramuscular es todavía más rápida (10-30 minutos). Si se precisa un efecto inmediato, se recomienda el músculo deltoides por su irrigación 3 veces superior a la musculatura glútea. Debido a su elevada fijación a las proteínas plasmáticas (90-98% del total), su molécula activa, es decir la fracción libre, aumenta considerablemente en situación de hipoproteinemia.
Se metabolizan en el hígado, basicamente en el sistema citocromo P-450. Su eliminación es urinaria y en menor proporción biliar. Con la edad el tiempo de eliminación aumenta, produciendo un mayor riesgo de acumulación del fármaco.
Alcanzan niveles plasmáticos estables en 5-10 días. Su vida media oscila entre 10 y 24 horas, facilitando así una cómoda administración de una sola dosis diaria cuando el paciente logra una condición estable.
Todavía persiste la controversia sobre la relación entre niveles plasmáticos de los antipsicóticos y su respuesta clínica. Los resultados hasta ahora obtenidos al respecto no son concluyentes. Tampoco parece definitiva la existencia de "ventanas terapéuticas" para los antipsicóticos.
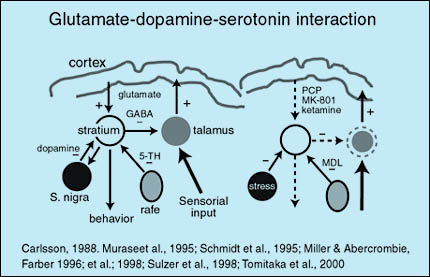
En los últimos años el estudio del sistema glutamatérgico ha adquirido mayor relevancia en la esquizofrenia. Las vías glutamatérgicas son parte de un sistema de interacciones neuronales que mantienen el equilibrio del funcionamiento de estructuras subcorticales. Las vías excitatorias glutamatérgicas que provienen de la corteza ejercen un efecto agonista a nivel del estriado sirviendo de contrapeso a las vías inhibitorias dopaminérgicas y serotoninérgicas que provienen de la sustancia nigra y el rafe dorsal respectivamente. Esta interacción permite la adecuada función del estriado el cual interactúa con el tálamo por medio de interneuronas GABAérgicas inhibitorias. El estriado es además muy importante en el control del comportamiento, mientras el tálamo es crucial en el control de los impulsos sensoriales provenientes de los órganos sensoriales. Si experimentalmente se administra un antagonista de receptores NMDA a voluntarios sanos, se producirá una predominancia de los impulsos inhibitorios en el estriado, provocando una disfunción de éste y el tálamo y conduciendo a alteraciones comportamentales y sensoriales propias de la esquizofrenia.
Fuente:http://psicofarmacologia.info/curso/esquizofrenia/Fig36.html
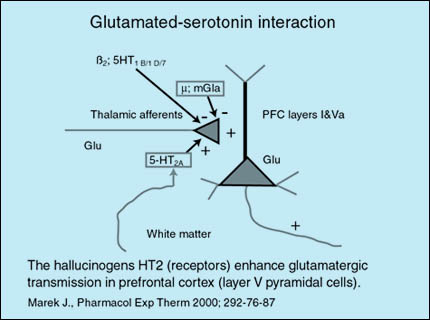
Las vías glutamatérgicas talamocorticales pueden ser moduladas además por proyecciones serotoninérgicas. En la corteza prefrontal (CPF) los receptores 5-HT(2A) facilitan la liberación de glutamato (Glu) de vías tálamocorticales excitatorias. Los receptores 5-HT(1B)(1D) y(7) por el contrario, inhiben la liberación de glutamato. Son también inhibitorios el receptor beta2 y el receptor glutamatérgico metabotrópico 2. En esquizofrenia, el disbalance dopamina-glutamato en estriado permite la liberación de las vías tálamo-corticales excitatorias. Algunos antipsicóticos de nueva generación antagonizan los receptores 5-HT(2A) llevando a una reducción en la liberación excesiva de glutamato a nivel cortical.
Fuente: http://psicofarmacologia.info/curso/esquizofrenia/Fig37.html
Modelos fisiopatológicos de la esquizofrenia; de dopamina a glutamato, de glutamato a GABA
RESUMEN
A pesar de los avances en el conocimiento de las bases biológicas de la conducta, los mecanismos neurobiológicos precisos involucrados en la esquizofrenia permanecen desconocidos. Como consecuencia de esto las terapias farmacológicas actuales descansan más sobre bases empíricas que sobre explicaciones fisiopatológicas. En el presente trabajo se propone un modelo de la esquizofrenia que podría ser de utilidad en el diseño de nuevas estrategias terapéuticas. Este modelo intenta integrar recientes hallazgos neuropsicológicos y de neuroimagen con lo que hoy sabemos respecto a la biología del desarrollo y plasticidad cerebral normal. Se propone que la esquizofrenia es una enfermedad del neurodesarrollo caracterizada por una neurotransmisión glutamatérgica inadecuadamente modulada a consecuencia de la disfunción de interneuronas GABAérgicas en múltiples regiones del cerebro. Anormalidades sutiles en el balance entre GABA y Glutamato explicarían los defectos en la cognición, la conducta social y la coordinación motora reportados en las etapas pre-psicóticas de la esquizofrenia. Más tarde en la historia natural de la enfermedad, estados hiperglutamatérgicos desencadenados por la incrementada neurotransmisión dopaminérgica propia de la peri-adolescencia y adultez temprana llevarían a la psicosis. Esta excesiva actividad glutamatérgica conduciría a su vez a las reducciones progresivas en sustancia gris y blanca observadas en recientes estudios prospectivos. En apoyo a esta hipótesis, se describen estudios propios y de otros laboratorios con pacientes esquizofrénicos, así como en un modelo animal de exposición intermitente a fenciclidina. Como corolario, drogas moduladoras de la neurotransmisión glutamatérgica, tales como acamprosato y lamotrigina, son propuestas como estrategias terapéuticas potencialmente utilizables en las etapas tempranas de la esquizofrenia.
Fig.1
Sinópsis Glutamato
Figura 1. Se señalan algunos de los genes posiblemente involucrados en el riesgo desarrollar esquizofrenia y como variaciones funcionales en éstos y probablemente muchos otros genes involucrados en la regulación de la excitabilidad y plasticidad neuronal provocarían sutiles anormalidades en el neurodesarrollo cerebral. Noxas ambientales tanto pre, como postnatales contribuirían a la expresión del fenotipo de disfunción cognitiva leve a moderada durante la infancia y la adolescencia que caracteriza a los sujetos en alto riesgo de desarrollar la enfermedad.
¿Qué proceso patológico causa el deterioro en la estructura y neuroquímica cerebral, así como en los rendimientos cognitivos de los pacientes de una manera que no parece responder a las terapias farmacológicas hoy disponibles? Al menos dos hipótesis no excluyentes entre si resultan plausibles. La primera es que la psicosis en sí misma, y/o los fenómenos prodrómicos que la preceden, involucra algún mecanismo neurotóxico.
La segunda es que los tratamientos antipsicóticos en uso comprometen el trofismo o la viabilidad neuronal. En apoyo a la hipótesis de un efecto neurotóxico de los primeros episodios psicóticos, estudios clínicos retrospectivos han mostrando que tratamientos antipsicóticos tempranamente instalados se asocian con un mejor pronóstico funcional de los pacientes, y que mientras más tarde éstos son instaurados mayor es la incidencia de refractariedad a tratamiento(23). Sobre la base de estos hallazgos se propuso que estrategias de diagnóstico y tratamiento temprano podrían mejorar el pronóstico funcional de esta enfermedad(24). Desafortunadamente, estudios prospectivos indican que terapias antipsicóticas precoces tienen poco o nigún impacto sobre la intensidad de los déficit emocionales y cognitivos de los pacientes(25,26). Si bien estos hallazgos han sido interpretados como pruebas en contra de la hipótesis neurotóxica de los primeros episodios psicóticos(25), una interpretación alternativa es que el efecto neurotóxico de hecho existe, pero no es prevenido por los fármacos convencionales debido a que éstos no corrigen el fenómeno patológico fundamental responsable de este deterioro o, peor aún, lo exacerban. En acuerdo con esta interpretación, un reciente estudio de seguimiento de pacientes de primer brote, randomizados a tratamiento con haloperidol u olanzapina, encontró que en ambos grupos hubo una reducción en el volumen de sustancia gris en la corteza prefrontal que alcanzó significación estadística sólo en el grupo tratado con haloperidol(27). En consistencia con estos hallazgos, significativas reducciones en el volumen de sustancia gris y blanca fueron reportadas en monos tratados durante un año con haloperidol y olanzapina(28). Asimismo, la exposición crónica tanto a haloperidol como olanzapina se asocia con reducciones en la expresión de BDNF (29,30), una neutrotrofina crítica para la estabilidad dendrítica y la viabilidad neuronal(31). Lo cual concuerda conestudios conductuales(32), electrofisiológicos(33) y moleculares(34) indicando que la señal dopaminérgica es fundamental para facilitar la plasticidad y trofismo neuronal requeridos para el aprendizaje normal, un efecto que, como veremos más tarde es en buena parte mediado por una potenciación de la neurotransmisión glutamatérgica. En consecuencia, y dado que todos los antipsicóticos conocidos hasta ahora ejercen un significativo efecto antidopaminérgico, que involucra probablemente una disminución global de la señal dopaminérgica, más allá de los receptores D2 (ver bloqueo en deporalización en referencia 2), no resulta sorprendente que tratamientos crónicos con estos fármacos resulten en efectos negativos para el trofismo neuronal. En consistencia con esta idea, la magnitud de los decrementos en la sustancia gris observados en pacientes tratados con haloperidol y olanzapina, se correlacionó positivamente con los déficit cognitivos de estos pacientes(27).
Fig.2
Ácido glutámico
Figura 2. La maduración de la neurotransmisión dopaminérgica durante la adolescencia contribuiría a intensificar las anormalidades en la regulación de la excitabilidad cortical que preceden a la psicosis. La emergencia de estados hiperglutamatérgicos exacerbados por dopamina explicaría los sintomas de ansiedad y desorganización conductual característicos de la etapas prodrómicas de la enfermedad. De nos ser tratados a tiempo, la liberación excesiva de glutamato durante este período llevaría a retracción dendrítica y atrofia neuronal. Drogas moduladoras de la neurotransmisión glutamatérgica, tales como acamprosato y lamotrigina, podrían ser útiles para revertir estos síntomas prodrómicos y evitar su progresión a la psicosis y el deterioro cognitivo.
Retracción dendrítica inducida por hiperactividad glutamatérgica; ¿el eslabón perdido?
Múltiples estudios sugieren que las pérdidas progresivas en la sustancia gris y blanca de la corteza cerebral de pacientes esquizofrénicos ocurren a expensas de una disminución de la conectividad intracortical(9). A nivel ultraestructural, esta conectividad disminuida parece tener su sustrato anatómico en una reducción del número de espinas dendríticas en las neuronas piramidales de la capa III(66), precisamente aquella que recibe la mayor parte de los axones que interconectan la corteza cerebral. Si bien hasta ahora no es posible afirmar con certeza si esta selectiva disminución en el número de espinas dendríticas juega un rol causal en la fisiopatología de la esquizofrenia, o es un mero epifenómeno, una posibilidad atractiva es que una disfunción dendrítica inducida por niveles excesivos de glutamato juegue un rol central en la cadena de eventos que llevan a la esquizofrenia crónica. En apoyo a esta hipótesis, estudios en preparaciones de hipocampo a distintos estadios en la maduración neuronal, muestran que es posible inducir pérdida de espinas dendríticas, sin muerte neuronal en condiciones de liberación excesiva de glutamato(67). Interesantemente, la vulnerabilidad a esta pérdida de dendritas depende del estado maduracional del tejido, lo cual parece particularmente relevante si se considera que los estadios incipientes de la psicosis esquizofrénica ocurren en coincidencia con un período de intensa remodelación sináptica, particularmente en la corteza prefrontal(68). Así, del mismo modo que las espinas dendríticas de neuronas piramidales en preparaciones de hipocampo sólo son eliminadas por exposición a glutamato durante un período crítico de su desarrollo, es posible que la eliminación de sinapsis en la corteza prefrontal durante la adolescencia también dependa de liberación de glutamato durante un período crítico de su desarrollo. De este modo, anormalidades sutiles en el equilibrio GABA/Glutamato que en etapas pre-adolescentes provocaban síntomas cognitivos y emocionales de intensidad leve a moderada, bajo el influjo de los cambios asociados a la adolescencia, particularmente en la actividad dopaminérgica(69), podrían llevar a la emergencia de estados hiperglutamatérgicos de intensidad aún mayor. En apoyo a esta hipótesis, una creciente literatura neurocientífica básica indica que la señal dopaminérgica ejerce un poderoso efecto potenciador de la neurotransmisión glutamatérgica a diversos niveles(70). Así, dada la irreversibilidad de los re-ajustes sinápticos producidos en esta etapa del desarrollo, es esperable que, de no actuar a tiempo, la pérdida de conectividad producida por esta hiperactividad glutamatérgica alcance un nivel crítico que conduzca a estados de deterioro cognitivo y emocional sobre los cuales sea difícil intervenir una vez superado este período crítico.
Por otra parte, dado que un cierto nivel de actividad glutamatérgica es fundamental para el trofismo neuronal(71), es posible que el drástico bloqueo de la neurotransmisión dopaminérgica inducido por los antipsicóticos disponibles lleve a atrofia neuronal por hipoglutamatergia prefrontal.
Moduladores bifásicos de la neuro-transmisión glutamatérgica; nuevas recetas para un viejo problema
De los estudios revisados hasta aquí revisada, algunos hechos parecen perfilarse con cierta claridad. Primero, existe un deterioro progresivo en las etapas incipientes de la esquizofrenia que tiene correlatos estructurales, neuroquímicos y, lo más importante, cognitivos y emocionales. Segundo, los fármacos convencionales no parecen ser capaces de evitar este deterioro y, por el contrario, probablemente lo agravan.
Tercero, múltiples estdios tanto clínicos como pre-clínicos sugieren que la emergencia de estados hiperglutamatérgicos en la corteza prefrontal podría jugar un rol patogénico central durante estadios tempranos de la psicosis esquizofrénica, o incluso en etapas pre-psicóticas de la enfermedad. Cuarto, los estados crónicos de la enfermedad podrían estar más relacionados con el tránsito de estados hiper a estados hipo-glutamatérgicos. Quinto, este tránsito podría estar mediado por pérdida de espinas dendríticas y atrofia neuronal provocados por la exposición repetida a niveles elevados de glutamato en la corteza prefrontal. Sustentando estas ideas, niveles elevados de glutamina han sido reportados en la corteza prefrontal medial de pacientes esquizofrénicos de primer brote(72), así como en adolescentes en alto riesgo(73), pero no en pacientes esquizofrénicos crónicos(74). Dada la naturaleza dinámica de los procesos que llevarían de un estado a otro, y la posibilidad de que regiones hipofuncionales coexistan con regiones hiperfuncionales en el cerebro de los pacientes, drogas con efectos bifásicos podrían ser particularmente útiles. De entre los candidatos posibles, acamprosato, un fármaco modulador de glutamato y GABA usado para la prevención de recaídas en pacientes alcohólicos, podría cumplir con estas condiciones. Es así como estudios pre-clínicos sugieren que este fármaco, por mecanismos que se desconocen, puede actuar como un antagonista glutamatérgico bajo condiciones de hiperactividad glutamatérgica, y como un agonista bajo condiciones de hipoactividad glutamatérgica
(75). Del mismo modo, estudios realizados recientemente en un ratón hiperglutamatérgico a causa de déficit en la recaptura de glutamato, indican que acamprosato normaliza los niveles de glutamato en los mutantes, sin afectar los niveles de este neurotransmisor en los ratones normales
(76).
Considerando el rol crítico jugado por la neurotransmisión glutamatérgica durante el neurodesarrollo y la cognición normal, drogas que normalicen la neurotransmisión glutamatérgica aberrante sin afectar la normal podrían tener un lugar de privilegiado en el tratamiento de pacientes adolescentes y adultos jóvenes, quienes tal vez se encuentran en una etapa del desarrollo cerebral en la cual demasiado glutamato es malo y muy poco también(71).
Fuente: http://www.scielo.cl/scielo.php?pid=S0717-92272005000400006&script=sci_arttext
Aunque parece que dicha hipótesis ha fracasado en el campo farmacológico
Fármacos glutamatérgicos para la esquizofrenia (Revisión Cochrane traducida)
Rol del glutamato
El glutamato es el neurotransmisor excitatorio más importante del sistema nervioso central, estimulando todas las neuronas de una forma no específica.
Es sintetizado a partir de glutamina por la enzima mitocondrial, glutaminasa y luego almacenada en vesículas hasta su liberación.
El glutamato liberado por las neuronas puede tomar 2 vías: a) una parte es recaptada en la membrana presináptica mediante mecanismos altamente específicos, b) gran parte es captada por los astrocitos, la mayoría del glutamato glial es convertido en glutamina, que es transportada a la hendidura sináptica donde es convertido nuevamente a glutamato.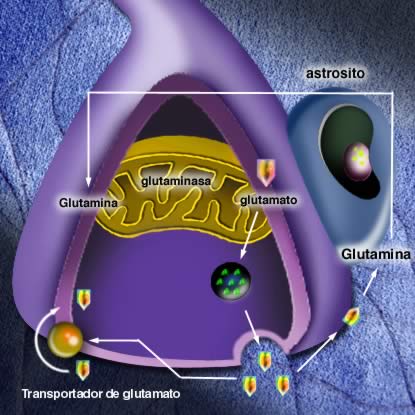
La liberación de glutamato en la hendidura sináptica es inhibida por drogas anticonvulsivantes como lamotigrina.
Hay 6 tipos de receptores postsinápticos de glutamato, 3 de ellos están unidos a canales iónicos, y los otros 3 son asociados a proteína G.
Los receptores asociados a canales iónicos son el N-metil-D-aspartato (NMDA), el a-amino 3-hidroxi 5-metilisoxazol 4-ácido propiónico (AMPA) y el receptor de Kainato. Los receptores asociados a proteína G son los receptores metabotrópicos tipo I, II y III.
El NMDA está ligado a un canal de calcio dependiente de voltaje. En estado de reposo no es activado por glutamato, pues un ion magnesio lo bloquea, cuando la membrana se despolariza, el magnesio se libera, permitiendo la entrada de calcio.
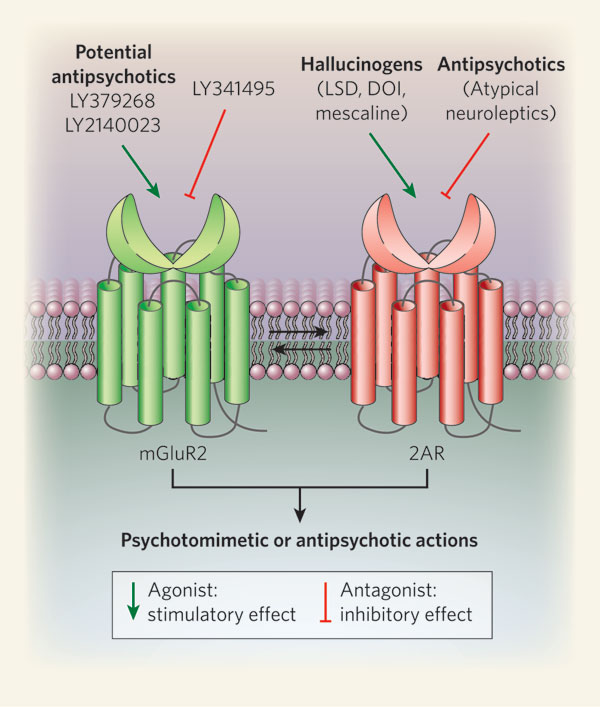 Este tipo de receptor se encuentra en todo el encéfalo, pero en mayores cantidades en el hipocampo y el neocórtex. Se ha asociado con procesos de aprendizaje y muerte neuronal cuando permanece despolarizado por períodos prolongados.
Este tipo de receptor se encuentra en todo el encéfalo, pero en mayores cantidades en el hipocampo y el neocórtex. Se ha asociado con procesos de aprendizaje y muerte neuronal cuando permanece despolarizado por períodos prolongados.
Anestésicos disociativos como la ketamina, y sustancias estimulantes como la fenilciclidina actúan a este nivel.
Se cree que los receptores NMDA tiene un rol importante en la patogénesis de la esquizofrenia, basados en la observación de los síntomas psicóticos producidos en casos de intoxicación por fenilciclidina. También se ha postulado que las alteraciones de la esquizofrenia son causadas por muerte neuronal acelerada mediada por estos receptores.
Los receptores de kainato se encuentran en cantidades abundantes en el hipocampo, están asociados a un canal de sodio que al permanecer abierto por tiempos prolongados, permite una entrada masiva del catión produciendo muerte neuronal por sobrecarga osmótica. En estudios de pacientes con esquizofrenia se han encontrado mayores concentraciones de este tipo de receptor en la corteza frontal, y disminución de los mismos en el hemisferio izquierdo.
Nuevos Antipsicóticos agonistas receptor mGluR2 antagonizan 2AR o receptor de la serotonia 5-HT2
http://www.nature.com/nature/journal/v452/n7183/fig_tab/452038a_F1.html
EFECTOS ADVERSOS DE LOS ANTIPSICOTICOS
NEUROLOGICOS----------- Distonía aguda. Acatisia. Parkinsonismo. Temblores periorales (Sdr. "del conejo"). Acinesia. Discinesia tardía. Disnea e Hiperventilación
GASTROENTEROLOGICOS--- Sequedad de boca. Sialorrea. Transtornos de la motilidad esofágica. Constipación. Ileo paralítico.
UROLOGICOS---- Retención urinaria. Tenesmo. Disuria. Polaquiuria.
HEPATICOS------ Colestasis intrahepática (ictericia). Hepatotoxicidad (clorpromacina).
CARDIOVASCULARES--------- Taquicardia. Hipotensión. Cambios ECG. Arritmias (tioridacina). * Algunas drogas antipsicóticas son bloqueantes de canales de calcio a nivel neuronal, del músculo cardíaco y del músculo liso. Por ejemplo, la tioridazina y la pimozida, lo cual puede explicar su mayor toxicidad cardíaca (prolongación del intervalo QT y taquicardia ventricular con riesgo de torsión de punta). Miocarditis en la clozapina por hipereosinofilia
HEMATOLOGICOS--- Leucocitosis. Eosinofilia. Aplasia medular. Trombocitopenia. Agranulocitosis o Neutropenia (clozapina) es la disminución aguda o crónica de granulocitos de la sangre, condición anormal de la sangre que puede predisponer al cuerpo humano a contraer infecciones.
ENDOCRINOS--------- Hiperprolactinemia. Amenorrea. Ginecomastia. Galactorrea (sulpiride). Aumento de peso.
Web interesante de los Orexígenos (estimuladores del apetito)
SEXUALES------- Transtornos de la eyaculación y/o erección. Pérdida de la libido. Frigidez.
DERMATOLOGICOS------- Dermatitis alérgica. Dermatosis por contacto. Fotosensibilidad. Urticaria. Decoloración de la piel. Pigmentación. Exantemas maculopapulares.
OCULARES------- Visión borrosa. Queratopatías. Cataratas estrelladas y planas (clorpromacina). Retinopatía pigmentaria (tioridacina). Empeoramiento del glaucoma.
S.N.C---------- Astenia. Sedación. Somnolencia. Disminución del umbral convulsivo. Delirium. Hipotermia (edad avanzada).
OTROS------- Síndrome Neuroléptico Maligno. Neumonía
Diabetes y sobrepeso
Diabetes tipo 2 y un gran aumento de peso, con causa multifactorial por bloqueo de diversos receptores a nivel central como periférico, verlo aquí
Una de las causas es que aumentan significativamente la actividad de la enzima AMP quinasa (AMPK) en el hipotálamo, región del cerebro que, entre otras funciones, regula el apetito por bloqueo de los receptores de la histamina H1. Estos dos elementos bioquímicos -el receptor de la histamina H1 y la enzima AMP quinasa- están relacionados con la regulación de la ingesta de comida.
Toxicidad Neuroléptica
Apoptosis neuronal
1. Toxicidad de los metabolitos de los antipsicóticos más en los típicos que en los atípicos, en las mitocondrias celulares produciendo muerte neuronal, concentración de monoaminas en el citoplasma e inhibición de la respiración celular mitocondrial por una selectiva inhibición de la NADH: ubiquinona oxirreductasa (en el complejo I).
2. Disminución de sustancia gris en el cerebro, retraso cognitivo, sensorial (retraso mental, retraso sensorial, además del motor) por disminución de la señal dopaminérgica Bloqueo D2 produce down regulation D1 en todo el cerebro e inhibiendo la producción de neutrofinas (plasticidad y desarrollo neuronal impidiendo apoptosis).
Por bloqueo receptores 5HT-2 y a1.
La familia de receptores 5HT2 está conformada por 3 subtipos de receptores ligados a proteína Gq, también el receptor a1, que estimulan el metabolismo de fosfatidilinositol Un fosfoinosítido, inositol fosfato o simplemente inosítido, es un fosfolípido que contiene en su estructura uno o más inositoles modificados por adición de uno o más grupos fosfato. Poseen especial relevancia en biología celular puesto que actúan como segundos mensajeros en la transducción de señal de las células.
Verlo con más detalle: en antidepresivos y en estimulantes
PROLONGACIÓN DEL INTERVALO QT
Torsades de Pointes
La medición correcta del intervalo QT en el electrocardiograma (EKG) se ha convertido en un parámetro muy importante, ya que numerosos medicamentos, tóxicos y enfermedades lo pueden prolongar y de esta manera favorecer la aparición de arritmias ventriculares malignas (Torsades de pointes) y muerte súbita. Esta revisión busca recordar algunos conceptos de fisiopatología, factores desencadenantes y tratamiento cuando nos enfrentamos a un paciente con prolongación del intervalo QT adquirido. El potencial de acción de la fibra ventricular o Purkinje resulta del movimiento transmembrana de iones a través de canales en la membrana celular. Gracias a este movimiento iónico la actividad eléctrica del corazón se ha dividido en dos grandes fases: la despolarización y la repolarización. La despolarización resulta del influjo neto de cargas positivas hacia en interior celular lo que estimula la contracción cardíaca y se representa en el trazado electrocardiográfico por el intervalo QRS; y la repolarización es el resultado de la salida de cargas positivas hacia el exterior celular con lo cual la célula miocárdica regresa al estado de reposo donde pueda ser nuevamente estimulada y esta representada en el EKG por el segmento ST y la onda T. El intervalo QT nos representa toda la actividad eléctrica ventricular, es decir tanto la despolarización como la repolarización ventricular. En el campo de la toxicología la prolongación del intervalo QT se ha evidenciado en la intoxicaciones por arsénico, cocaína, antidepresivos tricíclicos, fenotiazinas, fluoracetato de sodio y organosfosforados. Los protocolos de manejo tradicionales no consideran el monitoreo electrocardiográfico en los pacientes con intoxicación. Sin embargo existen varios reportes en la literatura, y lo hemos encontrado en nuestros pacientes, prolongación del Intervalo QT en individuos con intoxicación aguda, que antecede a la aparición de arritmias potencialmente fatales como la taquicardia ventricular polimorfa (torsades de pointes), la cual podría explicar la muerte súbita en pacientes aparentemente estables. Además han sido descritas otras anormalidades electrocardiográficas tales como: bradicardia o taquicardia sinusal, retraso en la conducción auriculoventricular o intraventricular, ritmo idioventricular, extrasistoles ventriculares polimorfas, taquicardia ventricular, fibrilación ventricular, prolongación de los intervalos PR, QRS; y cambios en el segmento ST. Todo esto enfatiza la necesidad de un monitoreo continuo de la actividad eléctrica cardiaca procurando determinar signos de mal pronóstico, y detectar tempranamente arritmias que pueden poner en riesgo la vida del paciente intoxicado.
Fuente
Fuente2 Efectos cardiovasculares Algunos antipsicóticos clásicos de baja potencia como la tioridacina se asocian a alteraciones de la repolarización cardíaca como la prolongación del intervalo QT. En pacientes predispuestos puede ser la causa de una muerte súbita. De entre los nuevos antipsicóticos, esta posibilidad sólo parece existir en el caso de la ziprasidona. Los antipsicóticos clásicos de baja potencia, así como los atípicos clozapina y risperidona, pueden producir hipotensión ortostática. Suele aparecer en los primeros días de administración y se desarrolla posteriormente tolerancia a este efecto.
Las taquicardias polimorfas sostenidas deben tratarse como fibrilación ventricular con desfibrilacion inmediata. Existe un tipo de taquicardia polimorfa con características muy especiales que se denomina Torsades de pointes, se encuentra una torsión de 180o del complejo QRS asociado a prolongación del Qtc y muchas veces es de origen iatrogénico (adquiridas), generalmente son autolimitadas y repetitivas. Las Torsades de pointes se han descrito como efecto secundario del uso de quinidina, sotalol, disopiramida, antidepresivos tricíclicos, tioridazina, eritromicina, pentamidina, trimetoprin sulfa, ampicilina espiromicina, terfenadina, astemizol, probucol, cocaína, adenosina y papaverina.
Fuente
SINDROME NEUROLÉPTICO MALIGNO
Se cree que el síndrome neuroléptico maligno se debe a un antagonismo sobre los receptores dopaminérgicos D2 en el hipotálamo, vías nigroestriadas y ganglios basales de la médula espinal. El resultado es la rigidez muscular y los temblores extrapiramidales. El bloqueo de los receptores hipotalámicos es la causa de la hipertermia. Además de estos efectos directos, el bloqueo de los receptores D2 puede producir el síndrome neuroléptico al eliminar la inhibición tónica del sistema nervioso simpático, con la correspondiente hiperactividad simpaticoadrenal. Por este motivo, los sujetos que muestren unos niveles basales de actividad simpaticoadrenal tienen un mayor riesgo de experimentar este síndrome.
Se ha demostrado que la dopamina es un neurotrasmisor esencial, especialmente en las patologías del SNC que incluyen control del tono muscular y de la termorregulación, precisamente las funciones que están alteradas en el SNM. La rigidez muscular, la akinesia, el mutismo y el temblor que caracterizan a este proceso pueden ser considerados como un desequilibrio dopaminérgico del hipotálamo53 Las anormalidades motoras son variadas, pero en general son típicas reacciones extrapiramidales parkinsonianas, comúnmente vistas como efectos colaterales de los neurolépticos (tabla 13). La administración de drogas tales como la bromocriptina , agonista de la dopamina, disminuye la severidad del SNM49 La hipertermia es producto del aumento de la producción endógena de calor y de la pérdida de la capacidad para disiparlo. El hecho de que el incremento de la temperatura sea proporcional a la rigidez sugiere, que la contractura muscular sea la responsable de la termogénesis. Una disminución de la contractura por acción de drogas tales como el dantroleno o la succinilcolina, disminuyen la temperatura. Sin embargo, experimentos en animales han establecido que un cambio en las concentraciones hipotalámicas de dopamina afectan a la termorregulación; así pues, el mecanismo por el cual se produce la hipertermia no está claro. Los trastornos electrolíticos han sido considerados un factor de riesgo para el desarrollo del SNM pues el proceso de recaptación de catecolaminas en las terminales nerviosas presinápticas es estrictamente dependiente de Na+; la hiponatremia aguda podría alterar las concentraciones de dopamina y noradrenalina en la hendidura sináptica, y por tanto favorecer la aparición del SNM, sobretodo en pacientes parkinsonianos54.
Fuente
http://www.ucm.es/BUCM/tesis/19911996/D/0/AD0067101.pdf
AUMENTO DE PROLACTINA
Las consecuencias de la elevación de la prolactina en pacientes que reciben drogas antipsicóticas son:
En mujeres: trastornos menstruales, galactorrea, congestión mamaria, disfunción sexual e infertilidad a corto plazo. A largo plazo producen disminución de la densidad ósea, mediada por una deficiencia relativa o absoluta de estrógenos, y probablemente también produzca enfermedad cardiovascular, cáncer de mama o endometrial y depresión. En hombres: A corto plazo producen disminución de la líbido, disfunción eréctil y eyaculatoria, disminución de la espermatogénesis y ginecomastia. A largo plazo producen disminución de la densidad ósea, mediada por una deficiencia relativa o absoluta de testosterona, y probablemente también produzca enfermedad cardiovascular, y depresión.
Fuente
DISNEA Y ACATISIA RESPIRATORIA
Conviene recordar también que las benzamidas sustituidas no son los únicos fármacos con capacidad para producir alteraciones respiratorias: la adenosina, los salicilatos, la medroxiprogesterona, la quetiapina según Shelton et al (10) y la olanzapina según Sattar y Gastfriend (11) están asociados a disnea e hiperventilación.
El mecanismo por el que estos medicamentos inducen hiperventilación no está aclarado por completo, pero se piensa que implica una acción directa sobre el centro respiratorio del cerebro medio así como una acción indirecta sobre los quimiorreceptores periféricos y neurotransmisores como la serotonina. Las complicaciones derivadas del síndrome respiratorio son la alcalosis respiratoria y la neumonía por aspiración. La sintomatología respiratoria puede estar relacionada con: Síndrome de Hiperventilación. Según Polkey et al (12) son varios los factores que pueden dar lugar a un Síndrome de Hiperventilación ya sean solos o por separado; dichos factores incluyen trastornos respiratorios tales como asma, enfermedad pulmonar intersticial o embolia pulmonar; la misma fiebre o la altitud y también la ansiedad, la depresión, la fobia y el trastorno de pánico (hiperventilación psicógena).
El síndrome de Hiperventilación se caracteriza por presentarse de forma súbita: respiración rápida y profunda (“hambre de aire”) con sensación de mareo, inestabilidad de la marcha, temblores, parestesias (sobre todo “hormigueo” peribucal y de los dedos) que pueden recordar a la tetania; disminución leve del nivel de conciencia (“atontamiento”, “presión craneal”); visión borrosa e incluso pérdida de visión; dolor de cabeza, náuseas, dolor torácico, taquicardia, malestar general; tinnitus. Otras manifestaciones como alucinaciones o síntomas somáticos (más comunes al lado izquierdo que al derecho) pueden conducir a diagnósticos erróneos tales como epilepsia, ataques isquémicos transitorios, enfermedades desmielinizantes o migraña.
La reproducción de los síntomas mediante la hiperventilación voluntaria se ha utilizado como prueba diagnóstica del síndrome de hiperventilación, aunque no es una prueba totalmente fiable. Según Phillipson (13) los pacientes con hiperventilación psicógena manifiestan, por una parte, disnea en reposo pero no durante el ejercicio leve y por otra parte, la necesidad de suspirar con frecuencia.
Acatisia Respiratoria Hirose (16) describe 5 casos que presentaron hiperventilación y disnea tan pronto iniciaron la toma de medicación antipsicótica y mejoraron al poco de suspenderla. Los pacientes se quejaban de tener una sensación interna de inquietud o malestar interno que les llevaba a buscar alivio mediante la respiración forzada, o dicho de otra manera, manifestaban la incapacidad de respirar de forma relajada (la misma incapacidad que otros pacientes manifiestan a la hora de controlar el movimiento de sus piernas). A este tipo de dificultad respiratoria Hirose la denominó “acatisia respiratoria” atribuyéndola al tratamiento neuroléptico mientras que su supresión y/o la administración de biperideno eliminaba o mitigaba la acatisia.
La acatisia respiratoria no siempre se acompaña de la inquietud motora típica y/o síntomas parkinsonianos, por lo que fácilmente puede pasar desapercibida. En nuestra opinión, la clínica descrita por Hirose es la que manifiesta la paciente del caso clínico, en donde lo más destacado es la incapacidad de respirar de forma sosegada, sin otros síntomas acompañantes. La suspensión del veralipride fue La discinesia respiratoria suele acom- pañarse de discine- sias faciales y orales y la dificultad respiratoria es continua durante las horas del día, disminuyendo o desapareciendo du- rante el sueño
Fuente
OTROS EFECTOS IATROGÉNICOS:
Efectos hematológicos
Se ha comunicado la aparición de leucopenias no graves y reversibles con muchos antipsicóticos. Sin embargo, la complicación potencialmente más grave es la aparición de agranulocitosis que se da con mayor frecuencia con la clozapina (aproximadamente 1%) por lo que se requiere la monitorización semanal del hemograma durante las primeras 18 semanas y posteriormente, de forma mensual mientras dure el tratamiento. Existe más riesgo durante el primer trimestre de tratamiento.
Efectos endocrinos El aumento de prolactina es más frecuente con los antipsicóticos convencionales que con los atípicos. Se produce por bloqueo dopaminérgico en el tracto tuberoinfundibular y, como consecuencia, puede provocar galactorrea y amenorrea en las mujeres y ginecomastia en los varones.
Efectos sexuales Hasta un 50 % de los pacientes tratados con antipsicóticos convencionales pueden sufrir disfunción sexual de algún tipo como disminución de la libido, anorgasmia o impotencia. Se han descrito casos de eyaculación retrógrada con la tioridacina y risperidona.
Efectos sobre el peso Todos los antipsicóticos pueden producir aumento de peso. Esto es más frecuente con los clásicos y con el atípico olanzapina.
Efectos dermatológicos Como con todos los fármacos se pueden presentar casos de dermatitis alérgica, más frecuentes durante las primeras semanas de tratamiento. Además, con algunos antipsicóticos convencionales como la clorpromacina se puede producir una reacción de fotosensibilidad tras la exposición al sol similar al eritema solar o incluso pigmentaciones.
Efectos oftalmológicos Un efecto potencialmente grave es el causado por la tioridacina cuando se administra a dosis superiores a los 800 mg/día. Puede producir este compuesto una pigmentación irreversible de la retina similar al de la retinosis pigmentaria. La clorpromacina puede provocar la aparición de opacidades en la córnea o conjuntiva sin afectar la visión.
Efectos hepáticos La administración de fenotiacinas como la clorpromacina puede producir una ictericia colestásica. Se observa con una frecuencia del 0,1% y aparece con más frecuencia durante los primeros meses de tratamiento. Una elevación transitoria y benigna de las transaminasas puede aparecer con la administración de algunos antipsicóticos atípicos como la olanzapina. Fuente
Enlaces:
6 meses para responder posts antiguos
¡Queremos tu opinión!
Si pregunta sobre este fármaco revise antes estos dos enlaces en particular
Ahora ya puede entender el prospecto y la ficha técnica


NEUROPSI













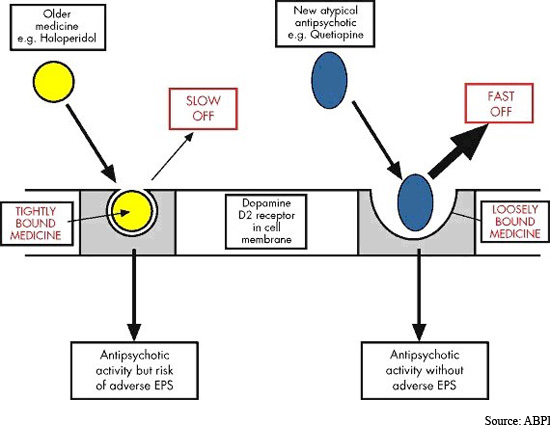
 Transducción de la señal
Transducción de la señal


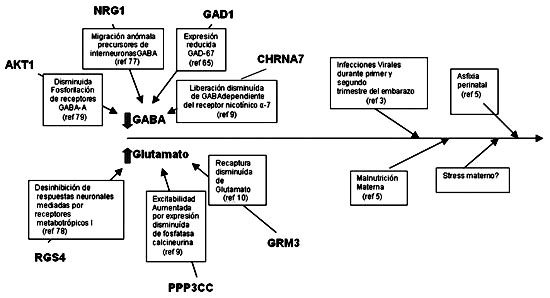{kind=link}
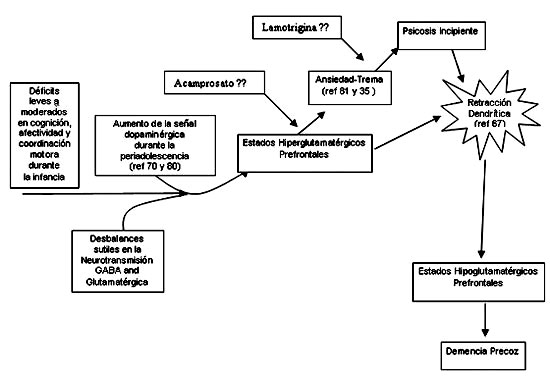{kind=link}
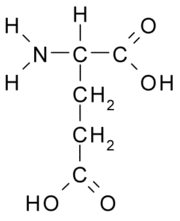{kind=link}